About this course:
This course reviews cystic fibrosis (CF), including relevant statistics, risk factors, genetic inheritance, pathophysiology, clinical features, diagnostic process, and evidence-based pharmacologic and non-pharmacologic treatment strategies.
Course preview
Cystic Fibrosis for APRNs
This course reviews cystic fibrosis (CF), including relevant statistics, risk factors, genetic inheritance, pathophysiology, clinical features, diagnostic process, and evidence-based pharmacologic and non-pharmacologic treatment strategies.
By the completion of this module, learners should be able to:
- Review relevant statistics regarding CF.
- Describe the risk factors, genetic inheritance, and pathophysiology of CF.
- Discuss the signs, symptoms, core features, and prognosis of CF.
- Describe the screening, testing, and diagnostic processes of CF.
- Discuss evidence-based guidelines for managing CF and treating complications, including medication therapy, prescribing indications, side effects, and monitoring parameters.
Cystic fibrosis is a rare genetic disorder characterized by chronic lung infections, progressive respiratory decline, and problems with digestion. Approximately 35,000 to 40,000 people in the U.S. and more than 100,000 worldwide are currently living with CF. While there is no cure for CF, advancements in its clinical management have led to significant improvements in life expectancy. Still, advanced cystic fibrosis lung disease (ACFLD), or end-stage lung disease, remains the primary cause of morbidity and mortality in patients who have CF. With the increasing number of adults who have CF, APRNs should be familiar with the condition, the trajectory of the illness, and the most up-to-date management guidelines (Centers for Disease Control and Prevention [CDC], 2024a; Kapnadak et al., 2020; National Heart, Lung, and Blood Institute [NHLBI], 2023c). In the interest of remaining inclusive, this activity will use the terminology caregiver to refer to parents, guardians, and other family members caring for pediatric patients who have CF.
Epidemiology
Approximately 1,000 new cases of CF are diagnosed each year, with more than 75% of affected persons diagnosed before their second birthday. CF was traditionally described as the most common life-threatening inherited disorder in white children, with an incidence of 1 in 2,500 live births. However, more recent epidemiological data demonstrate a shift in the demographics of affected populations. Currently, CF most commonly affects people of Northern European descent at a rate of 1 in 3,500. White children are still most prominently affected compared with other races. However, the incidence of CF is declining in most countries, and survival is increasing. First identified in 1938, infants diagnosed with CF typically die within their first year of life. Currently, more than 50% of people living with CF are 18 years and older. The Cystic Fibrosis Foundation (CFF) Patient Registry was founded in 1966 to track the health of patients who receive care at CFF-accredited care centers. Based on their 2023 Registry report, which includes data on people who have CF from 1986 to 2023, substantial changes in specialized CF care have improved survival. In 2023, there were 33,288 people who have CF in the Registry, with adults representing 60.4% of the CF population, compared with 31.1% in 1989. In 2023, patients who have CF had 100,648 clinic visits, 12,569 telehealth visits, and 10,164 hospitalizations. The annual mortality has also decreased to 7.0 per 1,000 people in 2023 compared to 16.6 per 1,000 people in 2003. Among people who have CF born between 2019 and 2023, 50% are predicted to live to over 61 years (CFF, 2023; Katkin, 2024a; Scotet et al., 2020; Sharma, 2024a).
Genes and Inheritance
CF is an inherited condition caused by a mutation in a specific gene that causes severe damage to the lungs, digestive system, and other organs in the body. Each cell in the body contains genetic information that provides coded instructions to make proteins, which determine how the body develops, looks, and works. Genes are found on thread-like structures inside the cell nucleus called chromosomes. Each chromosome comprises a strand of deoxyribonucleic acid (DNA) tightly coiled around histones (proteins) that support its structure. DNA contains the specific instructions required to perform necessary life functions, such as development, survival, and reproduction. In humans, each healthy cell contains 46 total chromosomes or 23 pairs. Genes are transmitted to offspring in pairs, with one copy inherited from each biological parent. Sometimes, genes can be inherited with mutations (i.e., abnormal changes or errors) in them, meaning they do not function properly (CFF, n.d.-b; Rogers, 2022; Scotet et al., 2020).
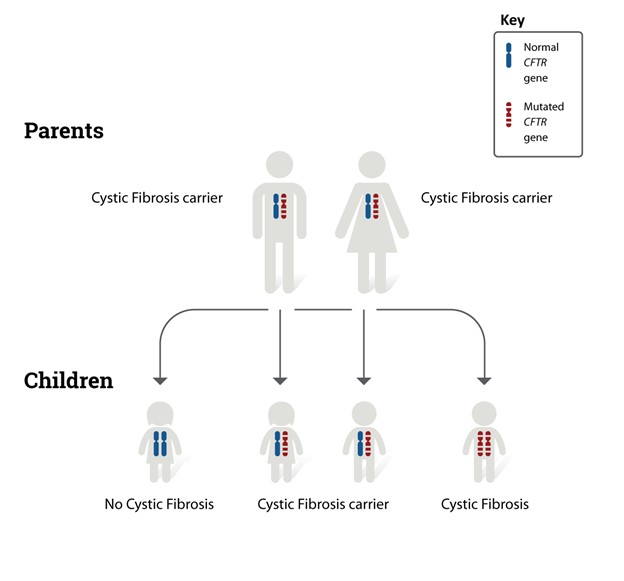
CF is transmitted via an autosomal recessive inheritance pattern (see Figure 1). Autosomal indicates that the gene is located on one of the first 22 pairs of chromosomes (i.e., those genes that do not determine gender); therefore, the disease affects males and females equally. Recessive means two copies of the gene (i.e., one from each biological parent) are necessary to inherit the disease. Therefore, the child must receive a copy of the cystic fibrosis transmembrane regulator (CFTR) gene mutation from each biological parent to inherit CF. Individuals who have CF inherit one copy of a genetic mutation of the CFTR gene from each biological parent. The CFTR gene mutation, located on the long arm of chromosome 7, causes the CFTR protein to malfunction. The CFTR gene is located in every organ within the body that makes mucus, including the liver, lungs, pancreas, intestines, and sweat glands. People who possess a single copy of a CFTR gene mutation are called “carriers” and are not affected by the condition. Carriers have no symptoms of the disease, but they have an increased chance of passing the defective gene to their children (CFF, n.d.-b; Sawicki, 2023). The American Lung Association (2024) reports that approximately 1 in 30 Americans are carriers of CFTR gene mutation. Each time two CF carriers have a child, the potential outcomes are as follows.
- 25% (1 in 4) chance the child will have CF.
- 50% (1 in 2) chance the child will be a carrier of CF.
- 25% (1 in 4) chance the child will not be a carrier of the gene and will not have CF (CFF, n.d.-b).
Figure 1
Autosomal Recessive Inheritance
(NHLBI, 2023b)
There are a few distinctions between the inheritance patterns for CF based on the biological parent’s status of affected, carrier, or non-carrier. Individuals who have CF can pass along copies of their CFTR gene mutations to their children. As shown in Figure 2, if an affected individual has a child who has a CFTR carrier, the outcomes are as follows.
- 50% (1 in 2) chance the child will be a carrier but will not have CF.
- 50% (1 in 2) chance the child will have CF (CFF, n.d.-b).
Figure 2
CF Affected Individual and Carrier Individual
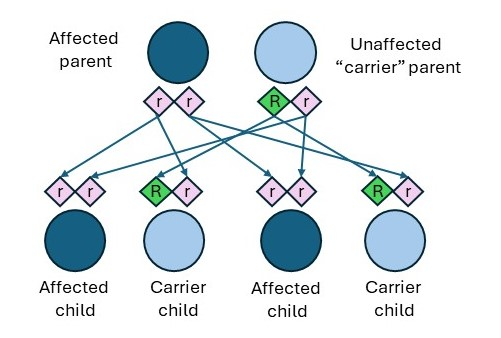
Two other inheritance patterns can lead to carrier children as follows (CFF, n.d.-b).
- If an affected individual has a child who has a non-carrier, there is a 100% chance that each child will be a carrier.
- If a non-carrier has a child who has a carrier, there is a 50% chance that each child will be unaffected and a 50% chance that they will be a carrier. There is a 0% chance of having an affected child.
While genetic inheritance is a known cause of CF, many children who have CF do not have a family history of the disease (i.e., the parents are both unawa
...purchase below to continue the course
Table 1
Impact of Ethnicity on the Risk of CF
Ethnicity | Risk of Being a Carrier | Risk of Having a Child Who Has CF |
White American | 1 in 29 | 1 in 2,500 to 3,500 |
Hispanic American | 1 in 46 | 1 in 4,000 to 10,000 |
African American | 1 in 65 | 1 in 15,000 to 20,000 |
Asian American | 1 in 90 | 1 in 100,000 |
(Cystic-Fibrosis.com, 2019; Stanford Children’s Health, n.d.)
Pathophysiology
The seventh pair of chromosomes contains the CFTR gene. Under normal conditions, the CFTR gene provides instructions for making the CFTR protein, which controls the movement of salts and fluids in and out of cells. Once the CFTR protein is created, it travels to the cell membrane, its primary function site. Here, the CFTR protein serves as an ion channel to maintain a balance between salt and water across cell membranes. It regulates the transport of negatively charged particles (chloride ions [Cl-]) and positively charged particles (sodium ions [Na+]) across cell membranes to produce a thin, freely flowing substance called mucus. Mucus is a slippery fluid that lubricates and protects the lining of the various organ systems, such as the airways, digestive system, and reproductive system. The CFTR protein is present on the surface of many epithelial cells, including those lining the airways, bile ducts, sweat glands, pancreas, sinuses, and vas deferens. When the CFTR gene is mutated, it does not work correctly and inhibits the transport of fluids and ions between cell membranes. Chloride becomes trapped in the cells, and water cannot hydrate the cellular surface, causing mucus build-up. CF mucus is dehydrated and viscous from defective chloride secretion and excess sodium absorption. As a result, the copious mucus that lines body cavities, especially the airways, becomes thick and sticky. This leads to mucostasis, as mucus plaques and plugs accumulate in the airway and other body surfaces (McKelvey et al., 2021; Rogers, 2022; Sharma, 2024b; Turcios, 2020).
The CFTR gene is extensive and complex, with more than 2,000 variants known to cause CF. This differentiation in variants is important because different mutations may affect which treatment options are available to patients. The variants are categorized into six classes based on how the mutation defect changes the functionality of the gene and correlates with disease severity. Classes I through III are associated with more severe disease manifestations and higher mortality. F508del, which is categorized as a class II defect, is the most common CFTR mutation, accounting for more than 70% of all CF cases. Classes IV through VI are linked to milder pulmonary disease. An overwhelming majority of affected persons who have CF have class I or class II mutations. A brief overview of each class is outlined in Table 2 (Brown et al., 2017; CFF, n.d.-b; Katkin, 2024b; Stanford Children’s Health, n.d.; Veit et al., 2016; Yu & Sharma, 2022).
Table 2
Classification of CFTR Mutations
Class | Defect Type | Description |
I | No functional CFTR protein (no protein)/protein production mutation | Nonsense mutations, splicing, or frameshift deletions result in severely reduced or absent CFTR expression. Class I dysfunction is caused by a premature termination of the messenger ribonucleic acid (mRNA) sequence, which fails to translate the genetic information (i.e., DNA). In other words, a functional CFTR is not created. Approximately 22% of people who have CF have at least one mutation in class I. |
II | Trafficking defect (no traffic)/protein processing mutation | Class II dysfunction results in abnormal post-translational processing of the CFTR protein, which is necessary for intracellular transport. As a result, the CFTR protein cannot be transported to the correct cellular location. The CFTR protein is created, but misfolding prevents it from moving to the cell’s surface, where it needs to perform the necessary functions. The most common CF mutation is F508del, which is primarily a protein processing mutation. This mutation removes a single amino acid from the CFTR protein that prevents it from staying in a normal 3-D shape. The cell recognizes that the protein is not the correct shape and disposes of it. Approximately 88% of people who have CF have at least one mutation in class II. |
III | Defective channel regulation (no function)/gating mutation | Class III dysfunction is characterized by diminished protein activity in response to intracellular signaling. The CFTR protein is created and moves to the cell’s surface but is non-functional. As a result, the channel gate does not open correctly to allow for the passage of fluids and ions. Approximately 6% of people who have CF have at least one mutation in class III. |
IV | Defective CFTR channel (less function)/conduction mutation
| Class IV dysfunction occurs when the protein is produced and correctly moves to the cell surface. However, the rate of chloride ion flow and the duration of channel activation after stimulation is decreased. In other words, the channel’s functioning is faulty. Approximately 6% of people who have CF have at least one mutation in class IV. |
V | Reduced synthesis (less protein)/insufficient protein mutation
| Class V dysfunction refers to the severely decreased protein concentration due to rapid degradation by cellular processes. This includes mutations that alter the stability of mRNA and the stability of the mature CFTR protein. As a result, the CFTR protein moves to the cell’s surface in insufficient quantities. Approximately 5% of people who have CF have at least one mutation in class V. |
VI | Decreased CFTR membrane stability (less stable) | The CFTR protein is created but has a decreased half-life and does not survive. |
(CFF, n.d.-h; Katkin, 2024b; Veit et al., 2016; Yu & Sharma, 2022)
The clinical consequences of the mutations outlined in Table 2 are decreased chloride secretion and increased sodium reabsorption into the cellular space. The increased sodium reabsorption leads to increased water resorption and the production of more viscous secretions from exocrine tissues. The mucus accumulates within nearly all organ systems, leading to obstructive pathologies. Exocrine glands secrete tears, sweat, milk, saliva, and digestive juices into a ductal system to an epithelial surface (e.g., the skin, gastrointestinal [GI] tract, or airways). Nearly all exocrine glands are affected by CF to some degree during their lifespan. The most commonly affected organs include the lungs, sinuses, pancreas, biliary and hepatic systems, intestines, and sweat glands. The glands become obstructed, malfunction, or produce excessive secretions that are extra sticky and thick. The sweat glands produce salty sweat, the lungs are more prone to infection, and the biliary and pancreatic ducts are blocked frequently. Sinus disease occurs when secretion viscosity increases and obstructs the sinus cavities. Over time, impaired sinus secretion clearance results in chronic sinusitis and secondary structural damage (Brown et al., 2017; Rogers, 2022; Veit et al., 2016; Yu & Sharma, 2022).
Complications
CF is a multiorgan disease with many associated complications, with the most common ones listed in Table 3 (Katkin, 2024a; Sawicki, 2023; Yu & Sharma, 2022).
Table 3
CF Impact on Multiorgan Systems
Body System | Complication/Manifestation |
Nose and sinuses |
|
Lungs |
|
Heart |
|
Liver |
|
Gallbladder |
|
Spleen |
|
Stomach |
|
Pancreas |
|
Intestines |
|
Reproductive |
|
Bone |
|
General |
|
(Katkin, 2024a; Sawicki, 2023; Yu & Sharma, 2022)
The most significant effect of CF is on the lungs, as respiratory failure is the leading cause of death. Although the lungs are usually normal at birth, most patients develop pulmonary disease during infancy or early childhood. The thickened mucus creates mucus plugs, clogs airways, impairs airway clearance, and facilitates the accumulation and colonization of bacteria. The abnormally thick mucus provokes the initial pulmonary injury in CF, as it causes diffuse obstruction within the bronchioles (i.e., small airways). Intermittent exacerbations with repeated infection and lung injury typically characterize the clinical trajectory of illness. Collectively, the disease results in a cascade of recurrent infections and chronic inflammatory processes. Mucus plugging in the bronchioles leads to obstructive lung disease, including bronchiectasis (i.e., abnormal dilation and damage to the bronchi), respiratory insufficiency, progressive airway destruction, and deteriorating pulmonary function (see Figure 3) (Brown et al., 2017; Katkin, 2024a; Sawicki, 2023).
Bronchiolitis is the most common lung infection in infancy and early childhood, causing inflammation and congestion within the bronchioles. In CF, chronic mucopurulent (i.e., secretions that contain both mucus and pus) plugging of the airways occurs secondary to obstruction and infection. The airways release proinflammatory cytokines, and repeated exacerbations lead to chronic inflammation and lung injury. Airway changes are more common than parenchymal changes, and about 50% of patients have bronchial hyperreactivity that can respond to bronchodilators. When the lungs become colonized with pathogenic bacteria, it becomes difficult to treat and eradicate the infections effectively, leading to clinical sequelae of multidrug-resistant bacteria colonization. Early in the disease course of CF, Staphylococcus aureus (S. aureus) is the most common pathogen. However, as the disease progresses, Pseudomonas aeruginosa (P. aeruginosa) is frequently isolated. In patients who have advanced pulmonary disease, chronic hypoxemia (i.e., low blood oxygenation) can lead to pulmonary artery enlargement, pulmonary hypertension, and right ventricular hypertrophy (Katkin, 2024a; Sawicki, 2023; Turcios, 2020).
In the GI tract, CF interferes with digestion; the pancreas, intestines, liver, and bile ducts are among the most commonly affected. When gastric contents pass into the duodenum, the pancreatic exocrine glands are triggered to excrete pancreatic enzymes into the small intestines to facilitate digestion—specifically, amylase (to digest carbohydrates), protease (to digest protein), and lipase (to digest fats). The mucus build-up of CF obstructs the pancreatic ducts, preventing the release of digestive enzymes, resulting in pancreatic insufficiency (PI) and maldigestion (i.e., the inability to digest food properly). The increased viscosity of secretions and obstruction of the pancreatic ducts inhibits the digestive process. In addition, decreased sodium bicarbonate composition lowers the gastric pH, impairing the breakdown of chyme. Chyme is a thick, semi-fluid mass of partially digested food and digestive secretions formed in the stomach and intestine during digestion. The abnormal acidity degrades the pancreatic enzymes before they reach the intestines, further preventing the digestion of food. When the chyme is not enzymatically processed appropriately, it leads to greasy stools, colicky abdominal pain, and malabsorption. Up to 95% of patients develop PI. In several of the most common CFTR mutations, the destruction of the pancreas may occur in utero; thus, the infant is born lacking the pancreatic enzymes. Patients with PI are incapable of absorbing essential nutrients, specifically fat-soluble vitamins (i.e., vitamins A, D, E, and K), which are notably deficient. Collectively, the destruction of pancreatic tissue, pancreatic duct obstruction, and insufficient enzymatic activity lead to malabsorption, malnutrition, and FTT (CFF, 2021a; Rogers, 2022; Sawicki, 2023; Turcios, 2020).
Intestinal involvement is also relatively common in CF. Roughly 15% of neonates who have CF present with meconium ileus at birth, an obstruction of the small intestines at the terminal ileum from abnormally thick and sticky meconium (feces). The cause of meconium ileus is multifactorial, but it is primarily due to increased fluid absorption due to the abnormal CFTR channel. The dehydrated intestinal contents cause constipation. In addition, some neonates can experience meconium plugging, in which the feces become enclosed in a mucus coat, making it more difficult to pass. Intestinal obstruction is expected later in life and can lead to inflammation, scarring, and stricture formation (Sabharwal & Schwarzenberg, 2023; Sawicki, 2023). Other common GI complications include the following.
- Intussusception (a form of bowel obstruction in which part of the intestines telescopes into an adjacent portion of the intestines)
- Volvulus (a loop of the intestine that twists around itself and the mesentery that supplies it)
- Rectal prolapse
- GERD
- Esophagitis (inflammation of the esophagus) (Sabharwal & Schwarzenberg, 2023; Sawicki, 2023)
Impaired glucose tolerance or diabetes mellitus (DM) occurs in about 2% of children. According to the American Diabetes Association (ADA, 2022), CF-related diabetes (CFRD) is the most common comorbid condition associated with CF, occurring in approximately 20% of adolescents and up to 50% of adults with CF. While the onset of CFRD is usually insidious, typically occurring between 18 and 24 years, autodigestion of the pancreas may occur as the pancreatic enzymes target the pancreatic tissues and result in pancreatitis. In the most severe cases, this can cause endocrine pancreatic failure when the pancreatic enzymes consume the islets of Langerhans. In most cases of CFRD, the underlying pathophysiology is related to the loss of function of the pancreatic islet cells, leading to insulin insufficiency. Other mechanisms include insulin resistance, progressive deterioration in glucose tolerance, delayed gastric emptying, altered intestinal motility, and liver disease. These patients require high caloric intake due to increased energy expenditure, malabsorption, and malnutrition. Compared to other populations who have diabetes, CFRD is associated with poorer nutritional status, more severe inflammatory lung disease, and higher mortality (ADA, 2022; Hameed & Robinson, 2023; Ode et al., 2022).
CF-related liver disease (CFLD) is another common complication, affecting approximately one-third of patients and causing significant morbidity and mortality. The term CFLD broadly refers to a spectrum of conditions affecting the liver in patients who have CF. The pathogenesis of CFLD is poorly understood since CFTR is expressed in the bile duct cells and gallbladder but not in the hepatocytes (i.e., the primary functional cells of the liver). Thickened CF mucus can irritate and block the bile ducts, preventing bile drainage out of the liver and gallbladder, leading to hepatic fibrosis (i.e., early-stage liver scarring) or cirrhosis (i.e., advanced or late-stage liver scarring). The liver becomes hard and nodular when it is scarred, causing increased pressure in the portal vein, enlarged blood vessels (varices), and splenomegaly (enlarged spleen). Approximately 30% of patients experience hepatic fibrosis, and 4% develop irreversible cirrhosis and portal hypertension by adolescence. Portal hypertension is the most significant form of CFLD and is typically related to cirrhosis. Additional hepatobiliary complications of CF include cholelithiasis (gallstones), affecting up to 15% of those who have CF, and cholecystitis (inflammation of the gallbladder) (Betapudi et al., 2023; Hercun et al., 2019; Leung & Narkewicz, 2024).
The sweat glands exhibit a distinct manifestation from all other tissues containing CFTR channels as the flow of chloride is reversed. Under normal conditions, sweat glands transport chloride from the extracellular space into the intracellular space, and the sodium and water are reabsorbed from the sweat gland tissues into the body. However, failure of the chloride channel to reabsorb chloride leads to sodium loss (excretion) onto the skin surface and subsequent fluid loss. These mechanisms cause the classical salty skin manifestation of CF. This is troublesome in warm environments or more severe cases, as there is an increased risk of hyponatremic dehydration (Brown et al., 2017; Sawicki, 2023).
In males, CF is known to cause infertility. The thickened mucus can block the vas deferens, the long tube that extends from the testes to the urethra. The epididymis is a series of tubes positioned posterior to the testicles where sperm are stored until they are made available at ejaculation. The vas deferens connect the epididymis to the ejaculatory ducts and serve as the canal to transport mature sperm through the penis during ejaculation. Nearly 95% of men who have CF are infertile because of an absent vas deferens, known as congenital bilateral absence of the vas deferens (CBAVD). In this condition, the sperm never reaches the semen, making it impossible to fertilize an egg through sexual intercourse. Less commonly, males may be diagnosed with obstructive azoospermia (OA), in which the spermatozoa are absent in the ejaculate despite normal spermatogenesis. In addition, females may have reduced fertility due to malnutrition and thickened cervical mucus, preventing conception (CFF, n.d.-e; Katkin, 2024a).
Signs and Symptoms
The symptoms of CF depend on which organ systems are affected and the severity of the disease. While CF has clinical implications across various organs, its most detrimental impact is on the lungs and GI tract (see Figure 3). The thicker and stickier mucus saturates the lungs, makes breathing and coughing more difficult and exhaustive, and heightens the risk of lung infections. People with milder forms of CF may have little to no symptoms, whereas others experience severe to life-threatening complications. As the condition advances with age, symptoms tend to evolve based on the impact and consequences on underlying organs. Because newborn screenings (NBS) are required across the U.S., CF is often diagnosed before symptoms develop. Typically, the earliest signs of CF in newborns and infants include meconium ileus, prolonged neonatal jaundice, or early lung infections. Young children may present with chronic cough, wheezing, anemia, or recurrent sinus and/or pulmonary infections. Others may present with poor weight gain, malabsorption in the GI tract, and failure to thrive (FTT). Boys may present with undescended testicles (Brown et al., 2017; Sawicki, 2023; Turcios, 2020). According to an analysis of 32,621 newborns, 31.2% were asymptomatic at the time of routine NBS. Among symptomatic infants, 33.1% demonstrated signs of acute or persistent respiratory abnormalities, 24.8% experienced FTT/malnutrition, 19.3% had steatorrhea (i.e., bulky, oily, foul-smelling stools) or otherwise abnormal stools, 16.3% endured a meconium ileus or other intestinal obstruction, 2.6% had electrolyte abnormalities, 3.7% had nasal polyps/sinus disease, and 2.4% suffered rectal prolapse (CFF, 2022). Adults who have CF commonly present with recurrent exacerbations of one or more affected organ systems. Table 4 lists some of the most common signs and symptoms of CF (Brown et al., 2017; Katkin, 2024a; Sawicki, 2023).
Table 4
Signs and Symptoms of CF
Generalized
|
Nasal/Sinus
|
Respiratory
|
GI
|
Genitourinary
|
(Brown et al., 2017; Katkin, 2024a; Sawicki, 2023)
Figure 3
Organs affected by CF
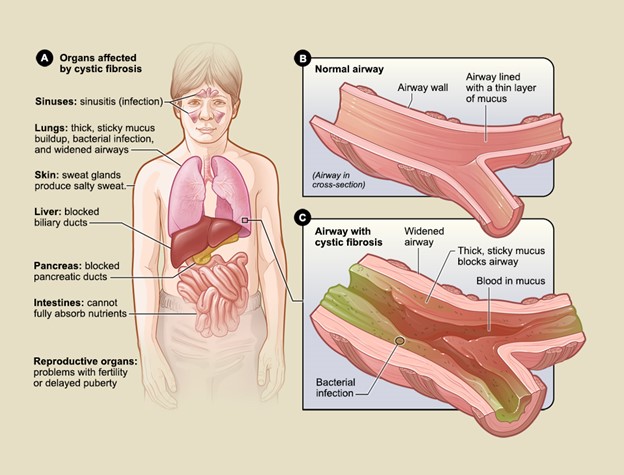
(NHLBI, 2023a)
Screening
Since 2010, NBS for CF has been required in all U.S. states. Some cases of CF are identified in utero during a prenatal ultrasound, which may demonstrate meconium peritonitis, bowel dilation, or absent gallbladder. These findings often lead to prenatal CF screening tests, although most patients are diagnosed through NBS following birth. While the particular screening protocol for CF may vary by state and institution, the screening algorithm is universally performed as a multi-step process. The NBS starts with testing for immunoreactive trypsinogen (IRT), an inactive precursor produced by the pancreas that is vital for breaking down the protein in food. In healthy people, trypsinogen is generated in the pancreas and transported to the small intestine, where it is converted to the enzyme trypsin to aid in digestion. In patients who have CF, mucus blocks the pancreatic ducts, preventing trypsinogen from reaching the small intestine. This obstruction causes hypertrysinogenemia or increased levels of trypsinogen in the blood. IRT is performed by collecting a blood sample via a heel stick using approved blood collection forms (i.e., Guthrie cards). There are strict criteria for heel sticks, including an optimal collection time between 24 and 48 hours after birth, precise collection, care, and air-drying procedures. The dried blood spot is also used to screen for many diseases aside from CF. Two IRT tests may be performed during the NBS (i.e., from the initial blood spot, and then a second one is collected about 2 weeks later). Approximately 20% of states routinely collect a second newborn blood specimen on every newborn in the U.S. The repeat testing practice is performed to demonstrate persistent hypertrysinogenemia. Further, monitoring the IRT can correlate with the severity of CF as it can decline below detectable levels, signifying the need to initiate pancreatic enzyme replacement (CFF, n.d.-d, n.d.-g; Farrell et al., 2017).
Diagnosis
Most cases of CF are first identified by NBS, although up to 10% are not diagnosed until adolescence or early adulthood. CF carrier screening is available in the U.S. and is recommended for couples planning pregnancy or seeking prenatal care. If both parents are carriers of a CFTR variant, prenatal screening of the fetus can be done by chorionic villus sampling (CVS) or amniocentesis. The wide phenotypic variability of CF can complicate prenatal counseling. A positive NBS does not confirm a diagnosis of CF; all positive NBSs must be followed up by a confirmatory sodium chloride (sweat) test. False-positive NBS results can occur for various reasons, such as misattributed or hemolyzed blood samples, medical errors in the laboratory analysis of the sample, labeling errors, or contamination with blood from other infants. Sweat testing is necessary for all patients with positive NBS, even those with two positive NBS samples. In addition, the sweat test verifies the physiologic abnormality found in CF (CFF, n.d.-d, n.d.-g; Farrell et al., 2017; Sawicki, 2023).
Despite advances in genetic testing, the sweat test is the universally recognized gold standard diagnostic test required to confirm a diagnosis of CF. The sweat test measures the amount of chloride in the infant’s sweat. As demonstrated in Figure 4, a small amount of a colorless, odorless chemical (e.g., pilocarpine) and electrical stimulation are applied to an area of the infant’s arm or leg. These areas are chosen to encourage the sweat glands to produce sweat. The sweat is collected and then analyzed in the laboratory to measure the quantity of chloride present in the sample. The sweat test is painless and usually takes about 30 minutes to complete (CFF, n.d.-d, n.d.-g; Farrell et al., 2017; Sawicki, 2023). The CFF has outlined the following recommendations regarding the sweat test.
- Sweat testing should be performed according to approved procedural guidelines, such as the Clinical and Laboratory Standards Institute (CLSI) guidelines.
- Sweat testing should be performed bilaterally when the infant weighs more than 2 kilograms (kg) and has a corrected gestational age of more than 36 weeks.
- Testing should be performed as soon as possible after the infant is at least 10 days old, ideally before 4 weeks of age.
- Sweat analysis should be performed within a few hours of collection (CFF, n.d.-d, n.d.-g; Farrell et al., 2017).
Figure 4
Sweat Test
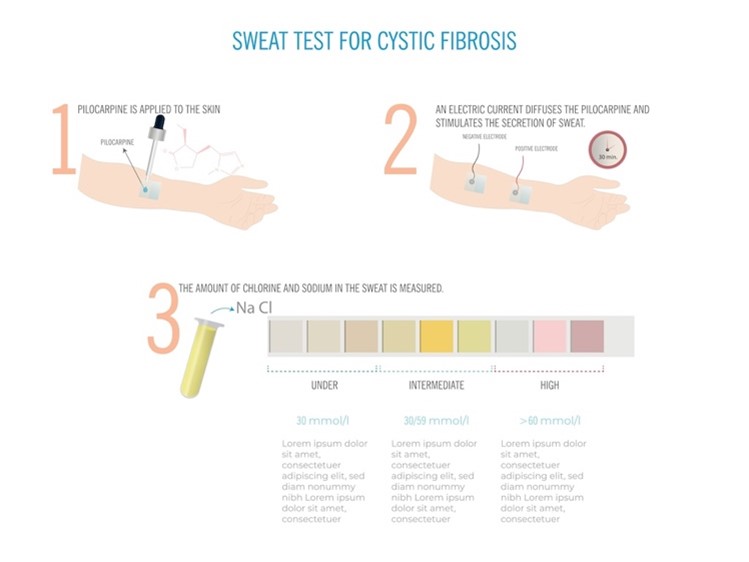
The possible results of the sweat test are outlined in Table 5 (CFF, n.d.-g; Farrell et al., 2017).
Table 5
Sweat Test Results
Chloride level | Interpretation |
Less than 30 mmol/L |
|
30-59 mmol/L |
|
≥ 60 mmol/L |
|
Quantity not sufficient (QNS) |
|
(CFF, n.d.-d, n.d.-g)
According to the CFF clinical care diagnosis guidelines (n.d.-d), if an infant is born with meconium ileus, a presumptive diagnosis of CF should be made while awaiting confirmatory testing results. In the presence of meconium ileus, the NBS can be falsely negative. However, meconium ileus usually occurs in the presence of severe pathogenic CFTR mutations. Other recommendations from the CFF include (Brown et al., 2017; CFF, n.d.-d, n.d.-g; Farrell et al., 2017):
- Individuals who have a positive NBS, clinical features consistent with CF, or a positive family history can be diagnosed with CF if the sweat test results are greater than or equal to 60 mmol/L.
- Individuals who have positive NBS and sweat test results greater than or equal to 60 mmol/L should undergo CFTR genetic testing if the CFTR genotype is not available through the screening process.
- CF is unlikely for individuals who have positive NBS and sweat test results of less than 30 mmol/L.
- CF is unlikely for individuals with clinical features that may be consistent with CF and have sweat test results of less than 30 mmol/L. A diagnosis could be considered if CFTR genotyping supports CF and not an alternative diagnosis.
- CF is possible for individuals who have a positive NBS, symptoms of CF, or a family history of CF with a sweat test result of 30 to 59 mmol/L on two different occasions. They should be considered for CFTR gene analysis.
- For individuals who have a positive NBS, symptoms of CF, or a positive family history, the identification of two CF-causing mutations is consistent with a diagnosis of CF.
- The absence of detection of two CFTR mutations does not exclude a diagnosis of CF.
- CF or a CF-related disorder should be suspected and considered in patients who have a normal sweat test but have the following characteristics that are suspicious for CF.
- A sibling who has CF
- A positive NBS
- Clinical symptoms consistent with CF in at least one of the following organ systems.
- Chronic sinus and/or pulmonary disease
- GI or nutritional abnormalities (including malnutrition and FTT)
- Salt loss syndromes
- CBAVD or obstructive azoospermia in males
For patients with inconclusive sweat tests and DNA testing results, a nasal potential difference (NPD) measurement can be used to evaluate CFTR dysfunction. NPD measurements are performed by placing electrodes in the nasal cavity and measuring voltage in the basal state. This is repeated after nasal perfusions with amiloride (Midamor) to block sodium transport and a chloride-free solution containing a cyclic adenosine monophosphate (cAMP) agonist (i.e., isoproterenol [Isuprel]) to stimulate the CFTR-dependent chloride transport. Patients who have CFTR dysfunction have a higher potential difference in the basal state, a greater decline following amiloride (Midamor), and minimal response to low chloride-isoproterenol (Isuprel) perfusion. CF can be diagnosed based on abnormal NPD. This test is not widely available and should only be performed in experienced centers. Additional diagnostic testing may be indicated depending on the presenting symptoms and the clinician’s judgment. For example, a plain chest radiograph (CXR) may help identify hyperinflation, bronchiectasis, abscesses, or atelectasis. Sinus radiography may demonstrate opacification of the paranasal sinuses. Abdominal imaging may be indicated in newborns who present with signs of meconium ileus (Brown et al., 2017; CFF, n.d.-d, n.d.-g; Farrell et al., 2017; Katkin, 2024a).
All infants with known or suspected CF should be tested for PI via fecal pancreatic elastase-1, a fecal test quantifying elastase. Elastase is an enzyme produced by the pancreas and excreted in the stool. Elastase is not broken down by other enzymes in the digestive tract and is eventually eliminated in the stool. Therefore, elastase can be detected and measured in the stool when the pancreas is functioning normally. The level in the stool is decreased when the exocrine tissues of the pancreas are not producing sufficient elastase and other digestive enzymes. Infants are diagnosed with PI if they have low fecal elastase (see Table 6). For accurate results, fecal elastase should only be obtained from stool from the rectum and not from an ostomy (if present). Fecal elastase collection should be deferred until the intestine is back in continuity if an ostomy is present. This screening test should be done on all patients at the time of diagnosis with CF. If the initial fecal elastase does not indicate PI, it should be repeated when the infant is older than 6 months and then annually. Screening is especially important in those patients at increased risk of PI based on the presence of two CFTR Class I, II, or III mutations (CFF, 2021a; Lab Tests Online, 2021).
Table 6
Fecal Elastase Results
Result | Interpretation |
Above 200 μg/g | Normal |
100-200 μg/g | Mild to moderate PI |
Below 100 μg/g | Severe PI (sensitivity/specificity of 100%) |
(CFF, 2021a; Lab Tests Online, 2021)
The ADA (2022) recommends annual screening for CFRD starting at age 10 in all patients who have CF not previously diagnosed with CFRD. Screening should be performed with the 2-hour, 75 g (1.75 g/kg) oral glucose tolerance test (OGTT). The glycosylated hemoglobin A1C (HbA1c) test is not recommended in this population due to its reduced sensitivity. Patients who have CFRD should be treated with insulin to attain strict glycemic goals. Annual monitoring for complications of diabetes should begin 5 years after the diagnosis of CFRD (ADA, 2022).
Management
CF is a systemic illness with broad implications for both quality and quantity of life when poorly controlled. There is currently no standard therapy for CF, and the condition is not curable. Management focuses on infection control and symptom management, intending to optimize function and maintain health for as long as possible. Treatment is geared toward preserving lung function by aggressively controlling respiratory infections, taking steps to thin secretions, clearing airways of mucus, and maintaining airway clearance. Emphasis should also be placed on optimizing nutritional status with pancreatic enzyme supplements and multivitamins. Given the high acuity level of these patients and their complex and multifaceted needs, they are best managed using a team approach incorporating specialists experienced in managing the condition and its complications. The ultimate goal is to preserve the quality of life and help patients optimize their functioning (Brown et al., 2017; Mogayzel et al., 2013; Sawicki, 2023; Simon, 2024c; Yu & Sharma, 2022).
Infection Prevention and Control (IPC)
Patients who have CF are more susceptible to infections and have a reduced capacity to recover, increasing the risk of fatal outcomes. Therefore, IPC strategies are the cornerstone of CF care. Specific guidelines outline the specialized precautions pertinent to this population. Adherence to IPC guidelines is encouraged to avoid infections, the transmission of CF pathogens, and the potential for pulmonary exacerbations. Last updated in 2013, the IPC clinical care guidelines were created by an interdisciplinary team and are endorsed by the CFF. The guidelines aim to help patients who have CF and their health care team reduce the risk of transmitting infectious micro-organisms. According to the guidelines, the following IPC strategies should be applied to all daily activities (Saiman et al., 2014; Simon, 2024c).
- All people who have CF should be separated by at least 6 feet (6-foot rule) from other people who have CF in all settings. This practice reduces the risk of cross-contamination or transmission of CF-prevalent pathogens between patients. Research has shown that patients who have CF can transmit dangerous micro-organisms and infections to each other, referred to as cross-infection. Cross-infection can lead to progressive illness, a decline in lung function, and death, depending on the situation. It is essential to recognize that the 6-foot rule does not apply to members of the same household.
- Only one person who has CF should attend any event, camp, educational retreat, or CF-sponsored indoor event or activity. However, family members who do not have CF and individuals who have CF and live together in the same household may attend these activities.
- All health care professionals, family members, friends, and close contacts of patients who have CF should regularly perform hand hygiene using an alcohol-based hand sanitizer or antimicrobial soap and water when hands could be potentially contaminated with pathogens.
- Smoking, vaping, second-hand smoke, and any other respiratory irritant exposure should be avoided.
- It is strongly recommended that all infants and children who have CF receive all the routine immunizations recommended by the American Academy of Pediatrics (AAP). This recommendation includes the following vaccinations.
- Measles, mumps, and rubella (MMR)
- Diphtheria-pertussis-tetanus
- Varicella
- Nirsevimab (Beyfortus) is recommended during the first respiratory syncytial virus (RSV) season for all infants who have CF.
- Haemophilus influenza (HIB)
- Prevnar (protects against common strains of Streptococcus pneumoniae [S. pneumoniae])
- COVID-19
- Annual influenza vaccine starting at 6 months of age
- If infants are younger than 6 months of age, all household contacts should be vaccinated against influenza.
According to the CFF (n.d.-f), the following IPC recommendations apply to health care settings and health care workers.
- Generate protocols, checklists, and audits to standardize safety and infection control practices when caring for patients who have CF.
- Disinfect surfaces before and after a person who has CF is in contact with them.
- Engage in routine hand hygiene. According to the CDC (2024b), proper hand hygiene is the most effective IPC risk reduction strategy. Table 7 provides detailed guidance on hand hygiene practices for health care professionals.
- Ensure that patients who have CF wear a surgical or isolation mask in all common areas, including hallways, elevators, and waiting rooms.
- Actively ensure patients who have CF always maintain a safe 6-foot distance in all settings.
- All health care professionals caring for patients who have CF should wear personal protective equipment (PPE), including gowns and gloves.
- Dedicate a single room to each patient who has CF.
- Evaluate patient activity on a case-by-case basis before advising a patient about participating in activities outside of a hospital room.
- Clean and disinfect nebulizers.
- Engage in respiratory hygiene/cough etiquette. The components of respiratory hygiene/cough etiquette include the following.
- Covering the mouth and nose during coughing and sneezing
- Using disposable facial tissues to contain respiratory secretions, with prompt disposal into a hands-free receptacle
- Wearing a surgical mask when coughing to minimize contamination of the surrounding environment
- Turning the head when coughing and staying at least 6 feet away from others, especially in common waiting areas
- Washing hands with soap and water or alcohol-based hand rub after contact with respiratory secretions
- Get vaccinated. Vaccination of health care workers and the public is one of the most prominent and compelling infection prevention and control strategies. The safest and most effective way to develop immunity and fight against a potential illness before it becomes dangerous is accomplished through vaccination. Vaccines work by impersonating the infectious agent, stimulating the immune system to generate antibodies against it without inducing the disease. If the pathogen subsequently attempts to invade the body, the body responds quickly, producing additional antibodies to combat the infection and thwart illness (CDC, 2023, 2024b; CFF, n.d.-f).
Table 7
CDC Hand Hygiene Guidelines
Health care workers should use an alcohol-based hand rub in the following clinical scenarios.
|
Health care workers should wash their hands with soap and water instead of using an alcohol-based hand rub in the following clinical scenarios.
|
Health care workers are advised to inspect their hands for breaks, cuts, or lacerations in the skin or cuticles before the start of each workday.
|
(CDC, 2024b, 2024c)
Respiratory Assessment
Monitoring lung disease progression is one of the most significant challenges in caring for patients who have CF. Airway inflammation, infection, and structural lung disease can begin during infancy and progress during early childhood. In addition, it is essential to have monitoring tools for children who are experiencing a new onset or an increase of symptoms and those who are experiencing a pulmonary exacerbation. Chest imaging may demonstrate evidence of regional or diffuse lung disease in CF. CXR remains the most frequently selected modality for monitoring lung disease. It is the most practical, is widely available, easy to perform, and uses a low level of ionizing radiation. However, CXRs in infants and young children may appear normal and may not necessarily correlate with presenting respiratory symptoms or functional impairment as seen clinically. Therefore, CXR should be obtained at least every 1 to 2 years to monitor disease progression in young children. More frequent monitoring with CXR should be considered in children with increased symptoms or those who fail to respond to appropriate treatment. Unfortunately, CXR imaging is limited; it is not sensitive enough to detect emerging small airway disease or early bronchiectasis. Chest computed tomography (CT) can more readily detect early changes in airway caliber, bronchial thickening, and air trapping and is the gold standard for diagnosing bronchiectasis. These changes may predate the development of respiratory symptoms, changes in physical exam, or lung function abnormalities. However, there are ongoing concerns about exposure to higher ionizing radiation, although protocols and technological advances have been developed to mitigate this risk. Further, infants and young children often require sedation or anesthesia to obtain acceptable CT images. For young children with CF, the routine use of chest CT to monitor the progression of lung disease is acceptable, but it is recommended that it be performed at less frequent intervals (every 2 to 3 years) and in place of a CXR. Abnormal findings on CT may correlate with early lung function abnormalities. Chest ultrasound is being increasingly used in acute pediatric settings when available. However, ultrasound images are limited in their assessment of airway caliber or thickness. Additionally, they cannot diagnose bronchiectasis or monitor the progression of structural lung disease in CF (Lahiri et al., 2016; Mogayzel et al., 2013).
PFTs are a vital tool for evaluating and monitoring lung disease and progression in CF and the most common method for longitudinal assessment. Spirometry is the most commonly used PFT and monitoring tool for pulmonary function in children and adults who have CF. With proper instruction and cooperation, spirometry can be performed successfully in children who have CF as young as 3 years old. Spirometry measures the forced vital capacity (FVC), the total volume of air exhaled during a forceful and complete exhalation after a maximal inhalation. The volume exhaled in the first second is known as the forced expiratory volume in 1 second (FEV1). The ratio between these two (i.e., FEV1/FVC) is the most important value reported from spirometry. These values allow for interpretation of the status of the lung ventilation function. They are compared to an expected normal for age, height, and gender. The measured value is calculated as a percentage of normal (e.g., normal equals 100%). A normal or high FEV1 with a low FVC may signify restrictive lung disease (e.g., interstitial lung disease, neuromuscular disease, pulmonary fibrosis). A low FEV1 with a high FVC indicates obstructive lung disease with airway trapping. Patients who have CF typically demonstrate air-trapping patterns with low FEV1 values proportional to the severity of the disease. PFTs will worsen from baseline during a pulmonary exacerbation or illness (Lahiri et al., 2016; Mogayzel et al., 2013).
Respiratory Management
Respiratory medications serve multiple purposes in CF. They are used to thin mucus, loosen secretions, kill bacteria, mobilize mucus to facilitate airway clearance, and lessen the work of breathing. The CFF (n.d.-c) recommends that chronic respiratory medications only be administered to patients 6 years and older, as there is insufficient evidence to support the use of chronic respiratory agents in children younger than 6 years. There are many options for respiratory support, including bronchodilators, antibiotics, anti-inflammatories, and mucolytics. Many respiratory medications are available as inhalers, nebulizers, or aerosol treatments. Bronchodilators such as albuterol (Pro-Air, Ventolin) and levalbuterol (Xopenex) are short-acting ß-agonists (SABAs) commonly used during pulmonary exacerbations. These agents selectively stimulate ß-2 adrenergic receptors, relaxing the airway smooth muscles, and are recommended for treating acute hyper-responsiveness. Their half-life is 2.7 to 6 hours, and they are available as metered-dose inhalers (MDI) or can be used in nebulizers. Bronchodilators dilate (open) the airways and allow for better penetration of other respiratory treatments. Although there is insufficient evidence of their effectiveness, CFF guidelines suggest using an SABA immediately before chest physiotherapy and exercise to facilitate the clearance of airway secretions. SABAs can also be administered before inhalation of nebulized hypertonic saline, mannitol (Osmitrol), or antibiotics to prevent non-specific bronchial constriction from these medications. Ipratropium bromide (Atrovent), another commonly prescribed respiratory medication, is a short-acting muscarinic antagonist (SAMA) that causes bronchodilation by antagonizing acetylcholine receptors. It has a half-life of 2 hours and is available as a nebulizer solution that can be mixed with albuterol (Pro-Air, Ventolin) or levalbuterol (Xopenex) or used independently as an MDI (Brown et al., 2017; CFF, n.d.-c; Mogayzel et al., 2013; Simon, 2024c).
Mucolytics work by thinning the mucus to help expectorate mucus from the lungs. Guidelines consistently recommend using mucolytics before performing pulmonary exercises to clear the airways. The two most commonly used mucolytics include inhaled hypertonic saline and dornase alpha (Pulmozyme). Hypertonic saline is a sterile solution of saltwater nebulized to facilitate mucus clearance by increasing the amount of salt in the airways, which attracts water (moisture) to the mucus, thinning it and making it easier to cough. Research demonstrates reduced lung infections in patients who have CF who use hypertonic saline twice a day. Dornase alpha (Pulmozyme) is a highly purified solution of recombinant human deoxyribonuclease (rhDNase), an enzyme that selectively cleaves DNA. In other words, it disassociates extracellular DNA strands to make the mucus thinner and looser. Splitting the DNA into shorter components helps break up thick mucus. Dornase alpha (Pulmozyme) is delivered to the lungs via a nebulizer and is indicated for use alongside standard therapies in patients aged 6 and older. According to clinical trials, daily use of dornase alpha (Pulmozyme) in patients with an FVC greater than or equal to 40% of predicted was shown to reduce the risk of respiratory infections requiring IV antibiotics. Therefore, the CFF (n.d.-c) recommends its use in patients aged 6 and older to improve lung function and reduce pulmonary exacerbations. Although it is generally well-tolerated, possible side effects include red and watery eyes, rash, rhinitis, loss of voice, throat discomfort, chest pain, and fever. Inhaled mannitol (Osmitrol) may be used as a second-line option to replace hypertonic saline for adults who do not tolerate or respond well to hypertonic saline or dornase alpha (Pulmozyme) (Brown et al., 2017; Cystic-Fibrosis.com, 2022a, 2022b; Mogayzel et al., 2013; Simon, 2024c; U.S. Food & Drug Administration [FDA], 2024).
Airway Clearance Therapy (ACT) is the backbone of preventative therapy in CF to preserve lung health for routine maintenance and during acute exacerbations. Daily ACT should be used in conjunction with medication therapy and all other pulmonary interventions offered. Airway clearance is critical for patients who have CF to remove mucus from their lungs and permit clean respiratory hygiene. Adequate airway clearance reduces the risk of lung infections and bacterial colonization. ACT techniques are vital as they help loosen and reduce mucus from the lungs. There are several different devices and methods used for ACT. Huff coughing is considered the underpinning of ACT. It includes inhaling, followed by a slow and strong exhale that is less forceful than full-blown coughing. It is credited with higher success since it is considered to be less tiring. Coughing should always be encouraged and never suppressed since it helps get rid of mucus. A typical cycle of 4 to 5 huff coughs is usually combined with other ACT techniques. Postural drainage and percussion (otherwise called chest physiotherapy) are commonly used in infants and young children. With this therapy, a therapist claps the patient’s chest and lower back simultaneously as they sit, stand, or lie in a position that helps loosen mucus. It usually involves alternating between lying in the left and right lateral, prone, and Trendelenburg positions. Research demonstrates that airway inflammation and obstruction exist even in infants; thus, it is recommended that airway clearance be started in the first few months of life. The “clapping method” is most commonly used for young infants to loosen secretions from the smaller airways. As children grow, there is the opportunity to use high-frequency chest wall oscillation devices, often referred to as the vest or oscillator. This technique uses an inflatable vest that wraps around the chest and attaches to a machine that vibrates at a high frequency. The vibrations help to loosen and thin the mucus and help facilitate airway clearance. Positive expiratory pressure therapy (PEP) is another commonly used technique during which a mask or mouthpiece attaches to a device called a resistor. The patient breathes in normally and breathes out against the resistance, which helps move air deeper into the lungs. There has not been a consensus statement to delineate which ACT method is the most useful. This decision should be based on patient preference, tolerance, and response. In addition to respiratory medications and ACT, multiple studies demonstrate the therapeutic benefits of regular exercise in patients who have CF to maintain and support lung function (Cystic-Fibrosis.com, 2022a; Lahiri et al., 2016; Simon, 2024c).
CFTR modulators
CFTR modulators are a novel group of medications that target the defective CF gene.
These therapies are designed to correct the genetic dysfunction by refining the CFTR protein’s production, intracellular processing, or functioning. Each medication targets a specific dysfunction caused by a gene mutation. CFTR modulators help correct the abnormal flow of electrolytes and fluids between the lungs, which helps to avoid thick mucus build-up. As more CFTR modulators have been developed and approved by the FDA, more recent recommendations suggest treating with the maximum number of modulators approved for the patient’s age group (triple therapy > dual therapy > monotherapy) (Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d).
Ivacaftor (Kayldeco)
Ivacaftor (Kayldeco) was FDA-approved in 2012 and was the first monotherapy CFTR modulator brought to market. It can currently be used for patients with a single mutation in the CFTR gene responsive to ivacaftor (Kayldec) based on clinical and/or in vitro assay data. Ivacaftor (Kayldeco) was developed to treat class III dysfunctions, where a mutation at G551D (gating mutation) is the primary abnormality, which occurs in 4.4% of patients who have CF. In vitro studies have shown that ivacaftor (Kayldeco) increases stimulated chloride flux for many other mutations besides G551D. The FDA has approved ivacaftor (Kayldeco) for 97 different mutations. Ivacaftor (Kayldeco) binds to the defective CFTR protein at the cell surface, restoring the proper function of the protein and opening the chloride channel. Ivacaftor (Kayldeco) was the first medication to directly impact the protein channel rather than treating the effects of CF. Reducing exacerbations, increasing FEV, improving lung function, and preserving overall lung health are strongly recommended (FDA, 2023a; Mogayzel et al., 2013; Simon, 2024d).
Dosing ivacaftor (Kayldeco) is age and weight-based for patients under 6 years. For infants who are 1 to 2 months old and greater than or equal to 3 kg, dosing is 5.8 mg orally every 12 hours. For infants 2 to 4 months old and greater than or equal to 3 kg, dosing is 13.4 mg orally every 12 hours. For children 2 months to 6 years old and 5 to 7 kg, dosing is 25 mg orally every 12 hours. Children 6 months to 6 years weighing 7 to 14 kg should take 50 mg by mouth every 12 hours, and those weighing more than 14 kg should take 75 mg by mouth every 12 hours. Dosing for all patients older than 6 years is 150 mg by mouth every 12 hours. Ivacaftor (Kayldeco) should be taken with fatty foods to increase absorption, although the optimal amount has not been established. The dose should be mixed with a small amount (1 teaspoon) of soft food or liquid if packets are used. When educating patients on this medication, APRNs should advise patients to avoid grapefruit and Seville oranges. These foods inhibit CYP3A4, the same enzyme responsible for ivacaftor (Kayldeco) metabolism. In addition, APRNs must perform a complete medication reconciliation before prescribing ivacaftor (Kayldeco) and counsel patients on the risk for harmful dietary supplementation and herbal interactions. Some of the most common medications and herbs that have significant drug interactions with ivacaftor (Kayldeco) include the following.
- Strong CYP3A inhibitors (e.g., itraconazole [Omnel, Sporanox], posaconazole [Noxafil], voriconazole [Vfend], ketoconazole [Nizoral], clarithromycin [Biaxin])
- Moderate CYP3A inhibitors (e.g., fluconazole [Diflucan], erythromycin [Erythrocin])
- Strong CYP3A inducers (e.g., rifampin [Rifadin], rifabutin [Mycobutin], phenobarbital [Luminal], carbamazepine [Tegrotol], phenytoin [Dilatin], and hypericum perforatum [St. John’s wort])
- CYP3A substrates (e.g., midazolam [Versed])
- P-gp substrate (e.g., digoxin [Lanoxin], cyclosporine [Neoral], tacrolimus [Prograf]) (FDA, 2023a; Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d)
Dose reductions are needed for patients who have hepatic impairment. Liver function tests should be monitored for patients on ivacaftor (Kayldeco). Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels should be evaluated at baseline and every 3 months for the first year, then annually after that. Because AST and ALT fluctuations are common in patients who have CF, managing liver enzymes on CFTR modulators can be complex. If the AST or ALT is greater than 5 times the upper limit of normal (ULN), holding the medication until values decrease is recommended. Once levels have normalized and the benefit outweighs the risk, the drug can be resumed. Ivacaftor (Kayldeco) also carries an increased risk of cataracts. Therefore, patients should undergo baseline and annual eye exams with an ophthalmologist. Other reported adverse effects (AEs) include headache, nasal congestion, oropharyngeal pain, upper respiratory tract infection, abdominal pain, diarrhea, nausea, rash, and dizziness. When selecting patients for ivacaftor (Kayldeco), each patient scenario should be considered. The potential for ivacaftor (Kayldeco) to prevent future lung damage should be considered, along with the risks for each patient (FDA, 2023a; Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d).
Lumacaftor/Ivacaftor (Orkambi)
Lumacaftor/ivacaftor (Orkambi) was FDA-approved in 2015 for use in children aged 1 to 2 years who are homozygous for the F508del mutation in the CFTR gene. They should be switched to elexacaftor/tezacaftor/ivacaftor (Trikafta) when the child turns 2. Lumacaftor/ivacaftor (Orkambi) is a “chaperone molecule” designed to transport the defective CFTR protein from the intracellular organelles, where it is processed, to the cell surface. When combined as lumacaftor/ivacaftor (Orkambi), it has been shown to have modest benefits for patients in improving PFTs, reducing the risk of pulmonary exacerbations, and increasing body mass index (e.g., weight gain). The BMI increase is beneficial in children who have CF, who are often underweight due to malnutrition and malabsorption of nutrients. Given the importance of weight in young children, lumacaftor/ivacaftor (Orkambi) may be an important addition to the treatment regimen in patients with poor weight gain. In addition, the potential to prevent long-term lung damage should be considered and weighed against the potential AEs of the medication (FDA, 2023b; Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d).
Lumacaftor/ivacaftor (Orkambi) dosing is based on weight. For children 1 to 2 years who are 7 to less than 9 kilograms, dosing is one 75/94-milligram packet orally every 12 hours. For children weighing 9 to less than 14 kilograms, dosing is one 100/125-milligram packet orally every 12 hours. The dose for children over 14 kilograms is one 150/188-milligram packet orally every 12 hours, with a max of four packets daily. Although it is recommended to change over to elexacaftor/tezacaftor/ivacaftor (Trikafta) at 2 years of age, lumacaftor/ivacaftor (Orkambi) can be continued with weight- and age-based recommendations. For children 2 to 5 years of age, recommended dosages mirror those described above for younger children. Children between the ages of 6 and 11 are typically given two 100/125-milligram tabs every 12 hours with a maximum of eight tabs daily. Most children over 11 and adults are maintained on two 200/125-milligram tabs by mouth every 12 hours. Before administration, each dose packet should be mixed with 5 milliliters of soft food or liquid at room temperature or below. After mixing the powder, it is stable as a mixture for 1 hour. However, the medication has a flour consistency and a bitter taste, making adherence challenging, particularly in children. APRNs should educate the caregiver on tips to mask the flavor of the drug. As with ivacaftor (Kayldeco), each dose should be taken with fatty foods to enhance absorption. Monitoring should include screening LFTs, but in addition to evaluating the AST and ALT, the bilirubin should also be assessed at baseline and every 3 months for the first year, followed by annually after that. If the AST or ALT is elevated greater than 5 times the ULN or 3 times the ULN and bilirubin is greater than 2 times the ULN, it is recommended to hold the lumacaftor/ivacaftor (Orkambi) until the values normalize (FDA, 2023b; Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d).
Lumacaftor/ivacaftor (Orkambi) carries a risk of adverse respiratory effects, including chest tightness and shortness of breath, which most commonly occur during the first few weeks of taking the medication. Since these symptoms may be difficult to detect in younger patients, APRNs should counsel caregivers to monitor for acute respiratory changes during early medication therapy. However, this is typically less common in younger patients than in older patients, which is potentially attributed to the less severe lung disease in children. Hypertension has also been reported, and therefore, it is recommended that blood pressure be monitored at routine clinic visits. Other reported AEs include upper respiratory tract infection, abdominal pain, diarrhea, rash, nausea, rhinorrhea, and cataracts. Unlike ivacaftor (Kayldeco), there are no foods to avoid with lumacaftor/ivacaftor (Orkambi). Because lumacaftor is a potent inducer of CYP3A, it inhibits and induces other CYP enzymes. Lumacaftor/ivacaftor (Orkambi) interacts with all forms of hormonal birth control, significantly reducing their efficacy. Therefore, female patients must be counseled on this risk once they reach puberty or at medication initiation. There are many other drug interactions with lumacaftor/ivacaftor (Orkambi), including the following.
- Strong CYP3A inducers (e.g., rifampin [Rifadin], rifabutin [Mycobutin], phenobarbital [Luminal], carbamazepine [Tegrotol], phenytoin [Dilatin], and hypericum perforatum [St. John’s wort])
- Sensitive CYP3A substrates with a narrow therapeutic index (e.g., midazolam [Versed], triazolam [Halcion])
- Immunosuppressants (e.g., cyclosporine [Neoral], everolimus [Afinitor], sirolimus [Rapamune], tacrolimus [Prograf])
- All CYP3A substrates (e.g., clindamycin [Cleocin], erythromycin [Erythrocin])
- All CYP2B6 & CYP2C substrates
- Acid suppressants (e.g., proton pump inhibitors [PPI] and histamine-2 blockers [H2])
- Antidepressants (e.g., citalopram [Celexa], escitalopram [Lexapro], sertraline [Zoloft])
- Corticosteroids (e.g., prednisone [Deltasone], methylprednisolone [Medrol]) (FDA, 2023b; Lahiri et al., 2016; Ren et al., 2017; Simon, 2024d)
Tezacaftor/Ivacaftor (Symdeko)
Tezacaftor/ivacaftor (Symdeko) yields modest improvement in pulmonary function and reduces the risk of pulmonary exacerbations for people with a homozygous F508del mutation, same as above for lumacaftor/ivacaftor (Orkambi). The F508del mutation interferes with CFTR protein folding and channel gating activity, and ivacaftor improves the gating abnormality while tezacaftor corrects the misfolding. Tezacaftor/ivacaftor (Symdeko) is FDA-approved for individuals who are 6 and older but has limited clinical utility when elexacaftor/tezacaftor/ivacaftor (Trikafta) is available. Tezacaftor/ivacaftor (Symdeko) is the drug of choice for people with one of five mutations approved for tezacaftor/ivacaftor (Symdeko) but not approved for elexacaftor/tezacaftor/ivacaftor (Trikafta). These individuals can start ivacaftor (Kayldeco) at 1 month of age and switch to tezacaftor/ivacaftor (Symdeko) at age 6. For individuals less than 30 kilograms, dosing is one 50/75-milligram tablet orally in the morning and ivacaftor (Kayldeco) 75 milligrams in the evening. For individuals greater than or equal to 30 kilograms, dosing is one 100/150-milligram tablet orally in the morning and ivacaftor (Kayldeco) 150 milligram in the evening. LFTs and bilirubin monitoring are recommended before and during treatment. Dose reduction is required for individuals with severe hepatic impairment, those taking P450 (CYP2A4)-inhibiting drugs, and those consuming foods containing grapefruit. Tezacaftor/ivacaftor (Symdeko) is well tolerated. The most common AEs are headache, nausea, sinus congestion, and dizziness (FDA, 2023c; Simon, 2024d).
Elexacaftor/Tezacaftor/Ivacaftor (Trikafta)
Elexacaftor/tezacaftor/ivacaftor (Trikafta) is a triple-drug combination for individuals over 2 years of age with at least one F508del mutation and individuals with any other CFTR gene mutation that is responsive based on in vitro or clinical trials. Approximately 92% of patients who have CF have a CFTR genotype that makes them eligible for elexacaftor/tezacaftor/ivacaftor (Trikafta). For individuals aged 2 to 6 years, weighing less than 14 kilograms, dosing is one 80/40/60-milligram packet taken orally in the morning and one 59.5-milligram ivacaftor (Kayldeco) packet in the evening. For those weighing more than 14 kilograms, the dosing is one 100/50/75-milligram packet taken orally in the morning and one 75-milligram ivacaftor (Kayldeco) packet in the evening. For individuals older than 6 and younger than 12 years of age, weighing less than 30 kilograms, dosing is two 50/25/37.5-milligram packets taken orally in the morning and one 75-milligram ivacaftor (Kayldeco) packet in the evening. For those individuals weighing greater than or equal to 30 kilograms or older than 12 years of age, the dosing is two 100/50/75-milligram packets taken orally in the morning and one 150-milligram ivacaftor (Kayldeco) packet in the evening. Elexacaftor/tezacaftor/ivacaftor (Trikafta) should be taken with fat-containing foods to improve absorption, and foods containing grapefruit should be avoided. Dose reductions are needed for patients who have hepatic impairment or those taking CYP3A4 inhibitors. Elexacaftor/tezacaftor/ivacaftor (Trikafta) is generally well tolerated, and the most common AEs are headache, upper respiratory infection, abdominal pain, diarrhea, rash, and nasal congestion (FDA, 2021; Simon, 2024d).
Lung Transplantation
Despite significant advancements in medical therapies for CF, the disease process continues to advance, and the lungs will ultimately fail from the disease process. Patients who have CF are estimated to live into their 40s before requiring lung transplantation for continued survival. Lung transplantation is the only definitive treatment for severe bronchiectasis, end-stage lung disease, and an FEV of less than 30%. The frequency of lung transplantation has declined with the introduction of CFTR modulators. The median survival following a lung transplant for children is 5.6 years; for adults, it is 9.9 years. The timing of the transplant is multifactorial, complex, and individualized. The International Society of Heart and Lung Transplantation (ISHLT) published consensus guidelines to guide the timely referral, assessment, optimization, and listing of potential lung transplant candidates. The intent is to help guide clinical decision-making when considering patients for lung transplantation based on multiple influences, recognizing that comorbidities and risk factors can negatively affect the post-transplant survival benefit. Because lung transplantation aims to improve survival and quality of life, historically, recommendations about allocating a scarce resource, survival benefit, were prioritized based on the ethical framework described in this document. A few priority criteria include the following: the 5-year predicted survival, baseline FEV1 value, the potential for development of pulmonary hypertension in the absence of a hypoxemic exacerbation, increasing frequency of exacerbations, and clinical decline. Since March 2023, the position of each candidate on the transplant list has been determined by a lung composite allocation score (CAS) formulated by the United Network for Organ Sharing (UNOS). The CAS is calculated using estimates of one-year survival without a transplant and five-year survival with a transplant. Nearly all lung transplant recipients who have CF will need a bilateral transplant since a native, diseased lung could be a source of infection that threatens the transplanted lung and possibly induces respiratory failure. It is important to note that transplantation is not a cure for CF, but it confers a prolongation of life and offers significant symptomatic relief (Leard et al., 2021; Mogayzel et al., 2013; Simon, 2023; Yeung et al., 2020).
Nutrition Therapy
All patients who have CF are encouraged to consume a high-fat, high-calorie diet with supplemental fat-soluble vitamins to compensate for malabsorption and help maintain a healthy weight. Oral feedings are preferred. However, if a patient’s intake does not meet metabolic demand, enteral feedings (e.g., gastric tube or jejunal tube) may be considered. Salt supplementation is also needed for infants who have CF, as the CFTR mutation in the sweat gland causes patients to excrete more salt compounds in their sweat than those without CF. Salt is also vital for normal growth and development. Those living in warm climates or participating in activities that cause excessive sweating are encouraged to consume additional sodium in their diet. Many will also require supplementation with water-soluble formulations of fat-soluble vitamins or CF-specific vitamins. Vitamin D supplementation is recommended in all exclusively breastfed infants and infants consuming less than 30 ounces of formula daily. Clinical practice guidelines also recommend measuring vitamin levels 2 months after starting the supplementation and then annually (Baker et al., 2023; Lahiri et al., 2016). Refer to the Nutritional Support in Critical Illness: Enteral and Parenteral Nutrition for APRNs NursingCE course for more information on nutritional therapy.
Most patients who have CF develop PI, as confirmed via the fecal pancreatic elastase-1. Ongoing, supportive therapy for patients who have PI includes pancreatic enzyme replacement therapy (PERT) with pancrelipase. PERT is one of the fundamental aspects of CF care as it replaces the digestive enzymes that are lacking. PERT should be prioritized and initiated as soon as PI is diagnosed, as dietary management is critical for healthy growth and development. PERT dosing often requires the skill of a qualified dietitian or nutritionist. The clinical guidelines regarding PERT dosing indicate that enzymes may be dosed based on grams of fat being consumed or based on weight. Adjusting the dosage of enzymes based on intake is more consistent with the body’s natural pancreatic enzyme production, while weight-based dosing is simpler and more convenient. Enteric-coated microencapsulated enzymes are the most effective, and patients should consistently use the same brand as they are not interchangeable. There are currently four oral pancreatic enzyme products approved for use in patients who have CF by the FDA: Creon®, Pancreaze®, Pertzye®, and Zenpep®. Enzymes should be taken at the beginning of every meal or snack that contains fat, such as dairy, meat, bread, and desserts (CFF, 2021a; Katkin et al., 2023; Lahiri et al., 2016).
Most formula-fed infants require 450 to 900 lipase units per gram of fat (units/g) or 2,000 to 4,000 units per 120 milliliters of formula. Breastfed infants typically require 2,000 to 4,000 units per feeding. Older children and adults often require 500 to 4,000 units/g or 500 to 2,500 units/kg/meal. Dosing should begin in the lower range, titrating up as needed. Patients should be encouraged to consume three meals and two to three snacks per day, with the PERT dosing cut in half for a snack. Excessively high doses of enzymes (greater than 6,000 units/kg/meal) increase the risk of fibrosing colonopathy, and it is unlikely that higher doses (greater than 4,000 units/g or 2,500 units/kg/meal) create a clinically significant improvement. Over-the-counter enzymes and those without an enteric coating are not recommended. Capsules may be opened and sprinkled into non-alkaline food but should not be crushed or allowed to sit for an extended period. Patients with inadequate enzyme activity typically report gastrointestinal symptoms (bloating, flatus, pain, and loose/frequent stools or steatorrhea) or poor growth/weight gain. Providers should assess patients for these symptoms consistently during regular follow-up visits, as well as evaluate dietary and adherence/compliance habits (CFF, 2021a; Katkin et al., 2023). Studies have shown that compliance with PERT has been as low as 27% to 50% (Barker & Quittner, 2016). Intestinal hyperacidity, abnormal intestinal motility, liver disease with low intestinal bile salt content, and several non-CF gastrointestinal conditions may also cause these symptoms and should be considered (CFF, 2021a; Katkin et al., 2023).
Pancreatic enzymes are generally well-tolerated, although some report initial abdominal pain, headache, diarrhea, flatulence, nausea/vomiting, or constipation. Fibrosing colonopathy is a known severe AE that should be discussed with patients and monitored closely. This complication is characterized by colonic strictures and is associated with high doses of PERT. Patients with fibrosing colonopathy typically present with evidence of obstruction, bloody diarrhea, ongoing diarrhea with abdominal pain and weight loss, or chylous ascites (i.e., the extravasation of chyle, a milky fluid rich in triglycerides, into the peritoneal cavity). Risk factors include patients under the age of 12, those taking more than 6,000 lipase units/kg/meal for longer than 6 months, and a history of meconium ileus, distal intestinal obstruction syndrome, or previous intestinal surgery. The diagnosis should be confirmed with advanced imaging studies or histopathology. If caught during the early stages, the condition may resolve with reduced enzyme dosing, although severe or advanced cases may require colectomy (CFF, 2021a; Katkin et al., 2023).
Psychosocial Support and Mental Health
Anxiety and depression are prevalent in patients who have CF and their families or caregivers. In the International Depression Epidemiological Study (TIDES), Quittner and colleagues (2014) evaluated depression and anxiety in patients who have CF and their caregivers in the U.S. and Europe. The study includes 3,000 mothers and 1,000 fathers of children who have CF, aged from birth to 18 years. The rates of depression in CF caregivers were triple those found in community comparisons, with 34% of mothers and 25% of fathers screening positive for depression. Anxiety was twice the rate of the community sample, with 48% of mothers and 36% of fathers screening positive for anxiety. If a mother was anxious, she was 15 times more likely to be depressed, whereas fathers were 9.2 times more likely to be depressed. Adolescent teens who have CF have twice the risk of depression or anxiety if either caregiver has depression or anxiety (Quittner et al., 2014). Research has demonstrated a link between the caregiver’s mental health, adherence, and health outcomes in children who have CF. Additional studies have shown that psychological symptoms in patients who have CF and their caregivers have been associated with decreased lung function, lower BMI, poorer health-related quality of life, more frequent hospitalizations, and increased health care costs. In addition, patients with symptoms of anxiety or depression have reduced participation in ACT, decreased pharmacological adherence, and poorer prognosis. To address the psychological burden of CF, the International Committee on Mental Health developed a consensus statement highlighting the critical importance of mental health screening and treatment in both patients and their caregivers. Coordinated efforts, screening, education, and supportive interventions to help encourage effective coping mechanisms and disease management skills can positively impact patient outcomes and improve mental and physical well-being (Barker & Quittner, 2016; Havermans & Willem, 2019; Quittner et al., 2014; Quittner et al., 2016).
Pulmonary Exacerbations
Because pulmonary disease is the most common cause of mortality in CF, it is vital to have a low threshold for diagnosis and intervention in pulmonary exacerbations. A pulmonary exacerbation denotes an acute worsening of lung function, commonly caused by an underlying infection. However, there is no universal definition of pulmonary exacerbation in any age group among patients who have CF. Still, they are often characterized by shortness of breath, fatigue, productive cough, chest congestion, and fever. Any acute respiratory illness should prompt admission to a hospital familiar with CF management. Pulmonary exacerbations should be managed with the following two priority objectives.
- Treat the underlying infection.
- Improve and maintain oxygenation (Lahiri et al., 2016; Mogayzel et al., 2013).
Pulmonary exacerbations are determined by the presence of cough in the majority of instances, but the differential diagnosis can be very broad (see Table 8). Pulmonary exacerbations can have variable presentations and can range from mild to severe. Some of the most common signs and symptoms include increased cough, change in sputum (e.g., color, volume, or consistency), new findings on respiratory examination (e.g., wheezing, crackles), increased respiratory rate, and increased work of breathing. In addition, patients may also experience fever, anorexia, and weight loss. Regardless of the underlying etiology, antibiotics to treat respiratory symptoms in all patients who have CF have consistently been associated with improved outcomes. Viral infections have been shown to increase the frequency of pulmonary exacerbations in children who have CF, may be associated with poor outcomes, and increase the risk of acquiring drug-resistant CF pathogens such as P. aeruginosa. The CFF (2021b) recommends continuing all of the patient’s regular maintenance respiratory medications and ACT interventions during pulmonary exacerbations. Anti-inflammatory medications such as glucocorticoids or corticosteroids (e.g., prednisone [Deltasone], dexamethasone [Decadron]) are used to relieve airway obstruction by reducing inflammation. These are used for a short duration and should not be prescribed prophylactically to prevent an exacerbation. Nasal cannula oxygen therapy should be prescribed to reduce the work of breathing. Bilevel positive airway pressure (BiPAP) ventilation may be required to overcome airway trapping in more severe cases. While intubation using mechanical ventilation is always an option, guidelines recommend reserving this for cases where respiratory failure is imminent. Ventilation and oxygenation should be supported with respiratory medications (Lahiri et al., 2016; Mogayzel et al., 2013; Simon, 2024a).
Table 8
Differential Diagnoses for Pulmonary Exacerbation in CF
|
(Brown et al., 2017; Lahiri et al., 2016; Mogayzel et al., 2013)
Studies demonstrate an evolution of pathogens in patients who have CF as they age. The most common pathogen in CF-associated lung infection in infancy and childhood is S. aureus, followed by Haemophilus influenzae (H. influenza). As the disease advances, adolescent and adult patients become colonized with increasing amounts of drug-resistant pathogens such as P. aeruginosa, Escherichia coli (E. coli), and Klebsiella pneumoniae (K. pneumoniae). Multidrug-resistant strains are more complex, generate more severe illness, and reduce treatment efficacy. For example, in the U.S., the prevalence of methicillin-resistant S. aureus (MRSA) in the respiratory tract is about 25%. Patients chronically infected with MRSA have a more rapid decline in pulmonary function and lower survival rates than those who are not (Sawicki, 2023; Turcios, 2020).
P. aeruginosa is the most common infectious etiology among adults who have CF. For this reason, guidelines include the use of broad-spectrum coverage against this pathogen. However, there are no consensus recommendations on what doses of certain oral antibiotics are most appropriate for patients who have CF. Guidelines strongly recommend obtaining a sputum culture and a sensitivity profile to identify all the pathogens present. Treatment recommendations are based on the most recent culture results, although in clinical practice, most clinicians will base antibiotic selection on the presence of organisms from multiple prior cultures. Guidelines recommend at least one antibiotic to cover each pathogenic bacteria cultured from respiratory secretions and two antibiotics for P. aeruginosa infections. Early detection and treatment of P. aeruginosa are vital to avoid the progression of CF lung disease and the increased risk of pulmonary exacerbations. Studies have demonstrated that failure to eradicate P. aeruginosa is associated with an increased risk of future pulmonary exacerbations (Lahiri et al., 2016; Mogayzel et al., 2013; Simon, 2024a; Turcios, 2020).
Antibiotics are a mainstay in CF care. Many patients who have CF take oral or inhaled antibiotics as part of their daily routines. Research demonstrates that oral antibiotics are the most prevalent treatment for pulmonary exacerbations. However, patients often require IV antibiotics (Stanford et al., 2021). Antibiotic selection and utilization evolve throughout life as the patient develops more resistant and difficult-to-treat lung infections. According to Mayer-Hamblett and colleagues (2018), a substantial reduction in pulmonary exacerbations was seen among children ages 6 months to 18 years treated with oral azithromycin (Z-Pak) plus inhaled tobramycin (TOBI) for early infection with P. aeruginosa. The researchers reported that the most significant reduction in pulmonary exacerbations was seen among children 3 years old and younger. Aztreonam (Azactam) is approved for children ages 7 and older with P. aeruginosa (CFF, n.d.-a; Lahiri et al., 2016; Mayer-Hamblet et al., 2018; Mogayzel et al., 2013; Simon, 2024a, 2024c). Inhaled antibiotics are commonly used to fight or control bacteria that cause lung infections in CF. Inhaled antibiotics are delivered directly into the small airways in the lungs and are most frequently used to improve respiratory symptoms in people with CF who have P. aeruginosa. According to CFF guidelines (n.d.-a), the most commonly used inhaled antibiotic selections include the following.
- Aztreonam inhalation solution (Cayston)
- Tobramycin (TOBI, Bethkis) inhalation solution
- Tobramycin (TOBI; Podhaler) inhalation powder (CFF, n.d.-a)
The 2013 CF pulmonary guidelines recommend prophylactic treatment with inhaled tobramycin (TOBI) for patients with persistent P. aeruginosa cultures to reduce exacerbations and maintain lung function. Oral azithromycin (Z-Pak) is only advised for patients with persistent P. aeruginosa cultures, not for patients with nontuberculosis mycobacteria (NTM), as there is evidence of resistance. Additionally, azithromycin (Z-pak) has anti-inflammatory effects that are therapeutic in patients with CF. Table 9 outlines some of the most commonly prescribed oral antibiotics to treat CF-related bacterial infections in pediatric and adult populations, respectively (Mogayzel et al., 2013; Muirhead et al., 2016). The CFF guidelines (2021b) state that there remains insufficient evidence to recommend an optimal duration of antibiotic therapy for acute pulmonary exacerbations. According to Stanford and colleagues (2021), antibiotic treatment typically lasts for 14 days but can range from 10 to at least 21 days in severe infections.
Table 9
Common Oral Antibiotic Regimens for Acute Pulmonary Exacerbations
Bacteria | Antibiotic | Pediatric Dose | Adult Dose |
S. aureus (methicillin-sensitive) | cefazolin (Ancef) | 100 mg/kg per day in 3 or 4 divided doses | 1.5 g every 6 hours or 2 g every 8 hours |
OR | |||
nafcillin (Nallpen) | 100 to 200 mg/kg per day in 4 or 6 divided doses | 2 g every 4 to 6 hours | |
Oral regimens for mild exacerbations (based on susceptibility)
| |||
S. aureus (methicillin-resistant) | vancomycin (Vancocin) | 60 mg/kg per day in 3 or 4 divided doses | 45 to 60 mg/kg per day in 3 divided doses |
OR | |||
linezolid (Zyvox) | Children <12 years: 30 mg/kg per day IV or orally in 3 divided doses Children ≥12 years: Use adult dose | 600 mg IV or orally every 12 hours | |
OR | |||
ceftaroline (Teflaro) | 45 mg/kg per day IV in 3 divided doses | 600 mg IV every 8 hours | |
Oral regimens (based on susceptibility)
| |||
One of the following: | |||
piperacillin-tazobactam (Zoysn) | 350 to 450 mg/kg per day in 4 divided doses | 4.5 g every 6 hours | |
ceftazidime (Fortaz) | 150 to 200 mg/kg per day in 3 or 4 divided doses | 2 g every 6 to 8 hours | |
cefepime (Maxipime) | 150 mg/kg per day in 3 divided doses | 2 g every 8 hours | |
imipenem-cilastatin (Primaxin) | 60 to 100 mg/kg per day in 4 divided doses | 0.5 to 1 g every 6 hours | |
meropenem (Merrem) | 120 mg/kg per day in 3 divided doses | 2 g every 8 hours | |
ticarcillin-clavulanate (Timentin) | 400 mg/kg per day in 4 or 6 divided doses | 3.1 g every 4 to 6 hours | |
PLUS | |||
ciprofloxacin (Cipro) | Oral dose: 40 mg/kg per day in 2 divided doses IV dose: 30 mg/kg per day in 3 divided doses | Oral dose: 750 mg every 12 hours IV dose: 400 mg every 8 to 12 hours | |
OR | |||
levofloxacin (Levaquin) | Oral and IV dose: 6 months to 4 years old: 16 to 20 mg/kg per day in 2 divided doses 5 to 16 years old: 8 to 10 mg/kg per day every 24 hours | Oral and IV dose: 750 mg every 24 hours | |
OR | |||
tobramycin (Tobrex) | 10 mg/kg every 24 hours (adjust dose for overweight patients) | 10 mg/kg every 24 hours (adjust dose for overweight patients) | |
OR | |||
amikacin (Amikin) | 30 to 35 mg/kg every 24 hours (adjust dose for overweight patients) | 30 to 35 mg/kg every 24 hours (adjust dose for overweight patients) | |
OR | |||
colistin (Coly-Mycin M) | 2.5 to 5 mg/kg per day in 3 divided doses | 2.5 to 5 mg/kg per day in 3 divided doses | |
S. aureus (methicillin-sensitive) and P. aeruginosa | Same antibiotics shown for P. aeruginosa alone EXCEPT that ceftazidime (Fortaz) should not be used because of poor activity against S. aureus | ||
S. aureus (methicillin-resistant) and P. aeruginosa | Same antibiotics shown above for P. aeruginosa alone Plus one of the following (3 antibiotics total) | ||
vancomycin (Vancocin) | 60 mg/kg per day in 3 or 4 divided doses | 45 to 60 mg/kg per day in 3 divided doses | |
OR | |||
linezolid (Zyvox) | Children <12 years: 30 mg/kg per day IV or orally in 3 divided doses Children ≥12 years: Use adult dose | 600 mg IV or orally every 12 hours | |
(CFF, n.d.-a, 2021b; Simon, 2024b)
Antibiotic-resistant infections are a priority concern for patients who have CF, as these can lead to fewer effective treatment options over time. Therefore, APRNs must counsel patients on the importance of taking the entire course of the prescribed antibiotic, including the proper sequence for taking the drug with other treatments and what to do if they miss a dose or cannot complete the entire course. The CFF guidelines recommend that inhaled antibiotics be used last, after the bronchodilator, mucolytics, and airway clearance techniques. This medication sequencing is important as it helps the antibiotics reach deeper into the lungs and provide more therapeutic benefits. Most studies in patients above the age of 6 have noted a reduction in pulmonary exacerbations requiring IV antibiotics in patients taking chronic inhaled CF medications (CFF, n.d.-a).
References
American Diabetes Association. (2022). Professional practice committee: Standards of medical care in diabetes- 2022. Diabetes Care, 45(Suppl 1), S1-S259. https://doi.org/10.2337/dc22-S002
American Lung Association. (2024). Learn about cystic fibrosis. https://www.lung.org/lung-health-diseases/lung-disease-lookup/cystic-fibrosis/learn-about-cystic-fibrosis
Baker, R. D., Baker, S. S., & Bojczuk, G. (2023). Cystic fibrosis: Nutritional issues. UpToDate. Retrieved September 11, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-nutritional-issues
Barker, D. H., & Quittner, A. L. (2016). Parental depression and pancreatic enzymes adherence in children with cystic fibrosis. Pediatrics, 137(2), e20152296. https://doi.org/10.1542/peds.2015-2296
Betapudi, B., Aleem, A., & Kothadia, J. P. (2023). Cystic fibrosis and liver disease. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK556086
Brown, S. D., White, R., & Tobin, P. (2017). Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. Journal of the American Academy of Physician Assistants, 30(5), 23-27. https://doi.org/10.1097/01.JAA.0000515540.36581.92
Centers for Disease Control and Prevention. (2023). Explaining how vaccines work. https://www.cdc.gov/vaccines/hcp/conversations/understanding-vacc-work.html
Centers for Disease Control and Prevention. (2024a). About cystic fibrosis. https://www.cdc.gov/cystic-fibrosis/about
Centers for Disease Control and Prevention. (2024b). Clinical safety: Hand hygiene for healthcare workers. https://www.cdc.gov/clean-hands/hcp/clinical-safety
Centers for Disease Control and Prevention. (2024c). Guideline for hand hygiene in healthcare settings (2002). https://www.cdc.gov/infection-control/hcp/hand-hygiene
Cystic-Fibrosis.com. (2019). How common is cystic fibrosis? https://cystic-fibrosis.com/statistics
Cystic-Fibrosis.com. (2022a). Types of airway clearance techniques. https://cystic-fibrosis.com/airway-clearance-techniques
Cystic-Fibrosis.com. (2022b). Types of mucus thinners used to treat cystic fibrosis. https://cystic-fibrosis.com/treatment/mucus-thinners
Cystic Fibrosis Foundation. (n.d.-a). Antibiotics. Retrieved September 11, 2024, from https://www.cff.org/Life-With-CF/Treatments-and-Therapies/Medications/Antibiotics
Cystic Fibrosis Foundation. (n.d.-b). CF genetics: The basics. Retrieved August 21, 2024, from https://www.cff.org/What-is-CF/Genetics/CF-Genetics-The-Basics
Cystic Fibrosis Foundation. (n.d.-c). Chronic medications to maintain lung health clinical care guidelines. https://www.cff.org/chronic-medications-maintain-lung-health-clinical-care-guidelines#chronic-medications-for-maintenance-of-lung-health-in-cf-patients-ages-6-and-older-executive-summary
Cystic Fibrosis Foundation. (n.d.-d). Clinical care guide for diagnosis of cystic fibrosis. Retrieved August 24, 2024, from https://www.cff.org/Care/Clinical-Care-Guidelines/Diagnosis-Clinical-Care-Guidelines/Clinical-Care-Guide-for-Diagnosis-of-CF.pdf
Cystic Fibrosis Foundation. (n.d.-e). Fertility in men with CF. Retrieved August 24, 2024, from https://www.cff.org/Life-With-CF/Transitions/Reproductive-Health-and-Fertility/CF-and-the-Male-Reproductive-System/Fertility-in-Men-With-CF
Cystic Fibrosis Foundation. (n.d.-f). Infection prevention and control clinical care guidelines. Retrieved August 24, 2024, from https://www.cff.org/medical-professionals/infection-prevention-and-control-clinical-care-guidelines
Cystic Fibrosis Foundation. (n.d.-g). Sweat test. Retrieved August 24, 2024, from https://www.cff.org/What-is-CF/Testing/Sweat-Test
Cystic Fibrosis Foundation. (n.d.-h). Types of CFTR mutations. Retrieved August 24, 2024, from https://www.cff.org/research-clinical-trials/types-cftr-mutations
Cystic Fibrosis Foundation. (2021a). Pancreatic enzymes clinical care guidelines. https://www.cff.org/Care/Clinical-Care-Guidelines/Nutrition-and-GI-Clinical-Care-Guidelines/Pancreatic-Enzymes-Clinical-Care-Guidelines
Cystic Fibrosis Foundation. (2021b). Pulmonary exacerbations clinical care guidelines. https://www.cff.org/pulmonary-exacerbations-clinical-care-guidelines
Cystic Fibrosis Foundation. (2022). 2022 patient registry annual data report. https://www.cff.org/medical-professionals/patient-registry
Cystic Fibrosis Foundation. (2023). 2023 Cystic Fibrosis Foundation patient registry highlights. https://www.cff.org/medical-professionals/patient-registry
Farrell, P. M., White, T. B., Ren, C. L., Hempstead, S. E., Accurso, F., Derichs, N., Howenstine, M., McColley, A. S., Rock, M., Rosenfeld, M., Sermet-Gaudelus, I., Southern, K. W., Marshall, B. C., & Sosnay, P. R. (2017). Diagnosis of cystic fibrosis: Consensus guidelines from the cystic fibrosis foundation. The Journal of Pediatrics, 181, S4-S15. https://doi.org/10.1016/j.jpeds.2016.09.064
Hameed, S., & Robinson, P. D. (2023). Cystic fibrosis-related diabetes mellitus. UpToDate. Retrieved September 3, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-related-diabetes-mellitus
Havermans, T., & Willem, L. (2019). Prevention of anxiety and depression in cystic fibrosis. Current Opinion in Pulmonary Medicine, 25(6), 654-659. https://doi.org/10.1097/MCP.0000000000000617
Hercun, J., Alvarez, F., Vincent, C., & Bilodeau, M. (2019). Cystic fibrosis liver disease: A condition in need of structured transition and continuity of care. Canadian Liver Journal, 2(3), 71-83. https://doi.org/10.3138/canlivj-2018-0019
Kapnadak, S. G., Dimango, E., H., Hadjiliadis, D., Hempstead, S. E., Tallarico, E., Pilewski, J. M., Faro, A., Albright, J., Benden, C., Blair, S., Dellon, E. P., Gochenour, D., Michelson, P., Moshiree, B., Neuringer, I., Riedy, C., Schindler, T., Singer, L. G., Young, D., … Simon, R. H. (2020). Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease. Journal of Cystic Fibrosis, 19(3), 344-354. https://doi.org/10.1016/j.jcf.2020.02.015
Katkin, J. P. (2024a). Cystic fibrosis: Clinical manifestations and diagnosis. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-clinical-manifestations-and-diagnosis
Katkin, J. P. (2024b). Cystic fibrosis: Genetics and pathogenesis. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-genetics-and-pathogenesis
Katkin, J. P., Baker, R. D., & Baker, S. S. (2023). Cystic fibrosis: Assessment and management of pancreatic insufficiency. UpToDate. Retrieved September 11, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-assessment-and-management-of-pancreatic-insufficiency
Lab Tests Online. (2021). Stool elastase. https://labtestsonline.org/tests/stool-elastase
Lahiri, T., Hempstead, S. E., Brady, C., Cannon, C. L., Clark, K., Condren, M. E., Guill, M. F., Guillerman, R. P., Leone, C. G., Maguiness, K., Monchil, L., Powers, S. W., Rosenfeld, M., Schwarzenberg, S. J., Tompkins, C. L., Zemanick, E. T., & Davis, S. D. (2016). Clinical practice guidelines from the Cystic Fibrosis Foundation for preschoolers with cystic fibrosis. Pediatrics, 137(4), e20151784. https://doi.org/10.1542/peds.2015-1784
Leard, L. E., Holm, A. M., Valapour, M., Glanville, A. R., Attawar, S., Aversa, M., Campos, S. V., Christon, L. M., Cypel, M., Dellgren. G., Hartwig, M. G., Kapnadak, S. G., Kolaitis, N. A., Kotloff, R. M., Patterson, C. M., Shlobin, O. A., Smith, P. J., Sole, A., Solomon, M., … Ramos, K. J. (2021). Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. The Journal of Heart and Lung Transplantation, 1-31. https://doi.org/10.1016/j.healun.2021.07.005
Leung, D. H., & Narkewicz, M. (2024). Cystic fibrosis: Hepatobiliary disease. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-hepatobiliary-disease
Mayer-Hamblett, N., Retsch-Bogart, G., Kloster, M., Accurso, F., Rosenfeld, M., Albers, G., Black, P., Brown, P., Cairns, A., Davis, S. D., Graff, G. R., Kerby, G. W., Orenstein, D., Buckingham, R., Ramsey, B. W., & Optimize Study Group. (2018). Azithromycin for early pseudomonas infection in cystic fibrosis: The OPTIMIZE randomized trial. American Journal of Respiratory Critical Care Medicine, 198(9),1177-1187. https://doi.org/10.1164/rccm.201802-0215OC
McKelvey, M. C., Brown, R., Ryan, S., Mall, M. A., Weldon, S., & Taggart, C. C. (2021). Proteases, mucus, and mucosal immunity in chronic lung disease. International Journal of Molecular Sciences, 22(9), 5018. https://doi.org/10.3390/ijms22095018
Mogayzel, P. J., Naureckas, E. T., Robinson, K. A., Mueller, G., Hadjiliadis, D., Hoag, J. B., Lubsch, l., Hazle, L., Sabadosa, K., Marshall, B., & Pulmonary Critical Practice Guidelines Committee. (2013). Cystic fibrosis pulmonary guidelines: Chronic medications for maintenance of lung health. American Journal of Respiratory Critical Care Medicine, 187(7), 68-689. https://doi.org/10.1164/rccm.201207-1160oe
Muirhead, C. A., Sanford, J. N., McCullar, B. G., Nolt, D., & MacDonald, K. D. (2016). Outpatient guide to management of pediatric cystic fibrosis acute pulmonary exacerbation. Clinical Med Insights in Pediatrics, 10, 57-65. https://doi.org/10.4137/CMPed.S38336
National Heart, Lung, and Blood Institute. (2023a). Cystic fibrosis symptoms. https://www.nhlbi.nih.gov/health/cystic-fibrosis/symptoms
National Heart, Lung, and Blood Institute. (2023b). What causes cystic fibrosis? https://www.nhlbi.nih.gov/health/cystic-fibrosis/causes
National Heart, Lung, and Blood Institute. (2023c). What is cystic fibrosis? https://www.nhlbi.nih.gov/health/cystic-fibrosis
Ode, K. L., Ballman, M., Battezzati, A., Brennan, A., Chan, C. L., Hameed, S., Ismail, H. M., Kelly, A., Moran, A. M., Rabasa-Lhoret, R., Saxby, N. A., & Craig, M. E. (2022). ISPAD clinical practice consensus guidelines 2022: Management of cystic fibrosis-related diabetes in children and adolescents. Pediatric Diabetes, 23(8), 1212-1228. https://doi.org/10.1111%2Fpedi.13453
Quittner, A. L., Abbott, j., Georgiopoulous, A. M., Goldbeck. L., Smith, B., Hempstead, S. E., Marshall, B., Sabadosa, K. A., Elborn, S., & the International Committee on Mental Health. (2016). Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax, 71, 26-34. http://dx.doi.org/10.1136/thoraxjnl-2015-207488
Quittner, A. L., Goldbeck, L., Abbott, J., Duff, A., Lambrecht, P., Sole, A., Tibosch, M. M., Brucefors, A. B., Yuksel, H., Castastini, P., Blackwell, L., & Barker, D. (2014). Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: Results of the international depression epidemiological study across nine countries. Thorax, 69(12), 1090-1097. https://doi.org/10.1136/thoraxjnl-2014-205983
Ren, C. L., Morgan, R. L., Oermann, C., Resnick, H. E., Brady, C., Campbell, A., DeNagel, R., Guill, M., Hoag, J., Lipton, A., Newton, T., Peters, S., Willey-Courand, D. B., & Naureckas, E. T. (2017). Cystic Fibrosis Foundation pulmonary guidelines: Use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Annals of the American Thoracic Society, 15(3), 271-281. https://doi.org/10.1513/AnnalsATS.201707-539OT
Rogers, J. L. (2022). McCance & Huether’s pathophysiology: The biological basis for disease in adults and children (9th ed.). Elsevier.
Sabharwal, S., & Schwarzenberg, S. J. (2023). Cystic fibrosis: Overview of gastrointestinal disease. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-overview-of-gastrointestinal-disease
Saiman, L., Siegel, J. D., LiPuma, J. J., Brown, R. F., Bryson, E. A., Chambers, M. J., Downer, V. S., Fliege, J., Hazle, L. A., Jain, M., Marshall, B. C., O’Malley, C., Pattee, S. R., Potter-Bynoe, G., Reid, S., Robinson, K. A., Sabadosa, K. A., Schmidt, H. J., Tullis, E., Webber, J., Weber, D. J., Cystic Fibrosis Foundation, & Society for Healthcare Epidemiology of America. (2014). Infection prevention and control guideline for cystic fibrosis: 2013 update. Infection Control & Hospital Epidemiology, 35(Suppl 1), S1-S67. https://doi.org/10.1086/676882
Sawicki, G. (2023). Cystic fibrosis. https://www.merckmanuals.com/professional/pediatrics/cystic-fibrosis-cf/cystic-fibrosis
Scotet, V., L’Hostis, C., & Ferec, C. (2020). The changing epidemiology of cystic fibrosis: Incidence, survival and impact of the CFTR gene discovery. Genes (Basel), 11(6), 589. https://doi.org/10.3390/genes11060589
Sharma, G. D. (2024a). Cystic fibrosis overview: Epidemiology. https://emedicine.medscape.com/article/1001602-overview#a5
Sharma, G. D. (2024b). Cystic fibrosis overview: Pathophysiology. https://emedicine.medscape.com/article/1001602-overview#a3
Simon, R. H. (2023). Cystic fibrosis: Management of advanced lung disease. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-management-of-advanced-lung-disease
Simon, R. H. (2024a). Cystic fibrosis: Antibiotic therapy for chronic pulmonary infection. UpToDate. Retrieved September 11, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-antibiotic-therapy-for-chronic-pulmonary-infection
Simon, R. H. (2024b). Cystic fibrosis: Antibiotic therapy for pulmonary exacerbations. UpToDate. Retrieved September 11, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-antibiotic-therapy-for-pulmonary-exacerbations
Simon, R. H. (2024c). Cystic fibrosis: Overview of the treatment of lung disease. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-overview-of-the-treatment-of-lung-disease
Simon, R. H. (2024d). Cystic fibrosis: Treatment with CFTR modulators. UpToDate. Retrieved August 30, 2024, from https://www.uptodate.com/contents/cystic-fibrosis-treatment-with-cftr-modulators
Stanford Children’s Health. (n.d.). The genetics of cystic fibrosis. Retrieved August 31, 2024, from https://www.stanfordchildrens.org/en/topic/default?id=the-genetics-of-cystic-fibrosis-90-P02933
Stanford, G. E., Dave, K., & Simmonds, N. J. (2021). Pulmonary exacerbations in adults with cystic fibrosis. Chest, 159(1), 93-102. https://doi.org/10.1016/j.chest.2020.09.084
Turcios, N. L. (2020). Cystic fibrosis lung disease: An overview. Respiratory Care, 65(2), 233-251. https://doi.org/10.4187/respcare.06697
U.S. Food & Drug Administration. (2021). Highlights of prescribing information: TRIKAFTA®(elexacaftor/tezacaftor/ivacaftor). https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212273s004lbl.pdf
U.S. Food & Drug Administration. (2023a). Highlights of prescribing information: KALYDECO®(ivacaftor). https://pi.vrtx.com/files/uspi_ivacaftor.pdf
U.S. Food & Drug Administration. (2023b). Highlights of prescribing information: ORKAMBI® (lumacaftor/ivacaftor). https://pi.vrtx.com/files/uspi_lumacaftor_ivacaftor.pdf
U.S. Food & Drug Administration. (2023c). Highlights of prescribing information: SYMDEKO® (tezacaftor/ivacaftor). https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/210491s011lbl.pdf
U.S. Food & Drug Administration. (2024). Highlights of prescribing information: PULMOZYME®
(dornase alfa). https://www.gene.com/download/pdf/pulmozyme_prescribing.pdf
Veit, G., Avramescu, R. G., Chiang, A. N., Houck, S. A., Cai, Z., Peters, K. W., Hong, J. S., Pollard, H. B., Guggino, W. B., Balch, W. E., Skach, W. R., Cutting, G. R., Frizzell, R. A., Sheppard, D. N., Cyr, D. M., Sorscher, E. J., Brodsky, J. L., & Lukacs, G. L. (2016). From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Molecular Biology of the Cell, 27(30), 424-433. https://doi.org/10.1091/mbc.E14-04-0935
Yeung, J. C., Machuca, T. N., Chaparro, C., Cypel, M., Stephenson, A. L., Solomon, M., Saito, T., Binnie, M., Chow, C., Grasemann, H., Pierre, A. F., Yasufuku, K., de Perrot, M., Donahoe, L. L., Tikkanen, J., Martinu, T., Waddell, T. K., Tullis, E., Singer, L. G., & Keshavjee, S. (2020). Lung transplantation for cystic fibrosis. The Journal of Heart and Lung Transplantation, 39(6), 553-560. https://doi.org/10.1016/j.healun.2020.02.010
Yu, E., & Sharma, S. (2022). Cystic fibrosis. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK493206