About this course:
This course examines the most common types of epilepsy and seizures across the lifespan, reviewing the incidence and prevalence, risk factors, clinical presentation, diagnostic criteria, and treatment modalities.
Course preview
Seizure Disorders and Epilepsy for APRNs
This course examines the most common types of epilepsy and seizures across the lifespan, reviewing the incidence and prevalence, risk factors, clinical presentation, diagnostic criteria, and treatment modalities.
Upon completion of this module, learners should be able to:
- identify the appropriate terms and definitions related to seizures and epilepsy
- consider the impact of epilepsy on healthcare in the US and worldwide
- describe the pathophysiology of seizures
- identify precipitating factors for seizures and epilepsy
- discuss the assessment and diagnosis of seizures and epilepsy
- discuss the management of a patient with seizures and epilepsy
- explore the self-management and education needed for patients with seizures and epilepsy
Epilepsy is a brain condition resulting in recurrent seizures. Seizures are disruptions in electrical communication between neurons. The term epilepsy is used if there are two or more unprovoked seizures (i.e., not caused by stimulation) more than 24 hours apart or after a single seizure if the patient is at high risk for further seizure activity (e.g., structural lesions such as a stroke, traumatic brain injury, or central nervous system [CNS] infection). Epilepsy is among the most common neurological disorders in the US and is associated with multiple conditions that affect mental and physical health. Epilepsy can shorten a person's life and significantly alter their well-being and ability to participate in activities of daily living (Adamolekun, 2022b; Centers for Disease Control and Prevention [CDC], 2020a; Schachter, 2022a). See Table 1 for key terms and definitions.
Table 1
Terms and Definitions
Term | Definition |
Seizure | An abnormal, sudden, excessive, uncontrolled discharge of neurons within the brain can result in a change in a person's level of consciousness (LOC), motor or sensory ability, or behavior. Seizures can occur as an isolated event or recur. The cause can be unknown, or there may be underlying conditions such as brain tumors or other illnesses; for example, diabetes may result in hypoglycemia and trigger a seizure. The term "features" is used to describe various signs and symptoms occurring during the actual seizure episode (Fisher et al., 2014; Ignatavicius et al., 2018). |
Absence seizure (petit mal seizure) | These are characterized by the lack of motor activity within the seizure. An absence seizure can mean an "absent stare," but a lack of motor activity can also be present in other seizure types (CDC, 2020b; Fisher et al., 2017). |
Epilepsy | Epilepsy is a disease of the brain defined by any of the following situations:
Epilepsy can be considered resolved in those with an age-dependent, self-limited epilepsy syndrome who are past the applicable age or have remained seizure-free for at least 10 years, without seizure medication for the past 5 years (Fisher et al., 2014). |
Aura | This sensation immediately preceding a seizure can be a smell, taste, visual, or a feeling of dizziness or numbness (Ignatavicius et al., 2018). |
Clonus | A spasm with continuous and repetitive patterns of rigidity and relaxation (Ignatavicius et al., 2018) |
Ictus | The actual seizure activity (Ignatavicius et al., 2018) |
Postictal | The period/state immediately after the seizure activity (Ignatavicius et al., 2018) |
Prodromal | This sensation occurs minutes to hours before an actual seizure. It is an early part of the seizure and may present as a headache or depressed feeling (Ignatavicius et al., 2018). |
Tonus | Part of the muscle contraction with excessive muscle tone (Ignatavicius et al., 2018) |
Reflex epilepsy | Seizures occur consistently in response to a stimulus or trigger (Epilepsy Foundation, n.d.-a). |
Unclassified seizure | A seizure occurs for no reason and does not fit into any generalized or focal classifications (Epilepsy Foundation, 2019a; Ignatavicius et al., 2018). |
Status epilepticus | A seizure lasting >30 minutes or ≥ two seizures without full recovery of consciousness between them. It is a medical emergency associated with significant morbidity and mortality (Ignatavicius et al., 2018). |
Epidemiology
Seizures commonly occur in children and adults, affecting 8% to 10% of the population over a lifetime. In addition, seizures account for 1% to 2% of all emergency department visits, with 25% of these visits being first-time seizures. Two-thirds of people who have a seizure never have a second. Epilepsy is the fourth most common neurological disorder worldwide, affecting approximately 65 million people. According to the CDC (2020b), approximately 1.2% of the US population has active epilepsy, equating to 3.4 million adults and nearly 470,000 children. There are 150,000 new cases of epilepsy in the US annually, and 1 in 26 people will develop epilepsy during their lifetime. Approximately one-third of those with epilepsy have uncontrollable seizures, with no current effective treatment modality. While many cases of epilepsy have known causes, over 60% have no known cause (Adamolekun, 2022b; Epilepsy Foundation, n.d.-b; Schachter, 2022a).
Types of Seizures
Seizures are grouped based on characteristics for clarity in clinical care, research, or education. Physicians, pharmacists, researchers, and other stakeholders can use the typing of seizures to refine treatment, determine medication efficacy, consider procedure eligibility, or determine the severity of a patient's diagnosis. Furthermore, consistent language among caregivers provides insight into each patient's condition and needs. The primary classifications of seizures are focal onset, generalized onset, and unknown onset. The origin of the seizure (i.e., where it starts in the brain) explains what may occur during the seizure and its projected effects, as well as the appropriate treatment course based on symptoms. Most seizures are classified as focal (i.e., the onset of electrical activity involves a focal region of the brain) or generalized (i.e., the onset of electrical activity involves both sides simultaneously). Focal seizures are then classified by level of...
...purchase below to continue the course
Focal-Onset Seizures
Focal-onset seizures originate within a hemisphere of the brain and may be localized or more widely distributed. For each seizure type, the beginning of the ictal phase (i.e., middle of the seizure) is consistent between seizures. They may exhibit specific patterns that involve the ipsilateral (i.e., same side) and/or contralateral (i.e., opposite side) hemisphere. Signs and symptoms during a seizure aid in identifying the specific area, lobe, or hemisphere involved in the seizure onset and further dissemination (Adamolekun, 2022b; ILAE, 2022e).
Focal seizures are first classified according to the level of awareness, which is critical to determine due to the impact on daily life functioning and safety. If a focal seizure does not impair awareness, it is classified as a focal aware seizure (formerly known as a simple partial seizure). If a seizure includes impaired awareness (patient loses awareness of their surroundings), it is classified as a focal impaired awareness seizure (formerly known as complex partial seizures). The initial sign/symptom is the most important as it identifies the region of the brain the seizure originates, even if the seizure has other features (Adamolekun, 2022b; ILAE, 2022e).
Focal seizures can be further classified by motor or non-motor onset:
- focal-onset motor seizures can include:
- atonic (focal loss of muscle tone)
- automatisms (coordinated, purposeless, repetitive motor activity)
- clonic (focal rhythmic jerking)
- epileptic spasms (focal extension or flexion of arms or flexion of trunk)
- hyperkinetic (causing thrashing or pedaling)
- myoclonic (brief, irregular focal jerking)
- tonic (sustained focal stiffening of a limb or a side of the body)
- focal-onset non-motor seizures are further classified based on the first prominent feature:
- sensory dysfunction (sensation present at onset such as tingling, flashing of lights, buzzing, odor, taste, dizziness, hot/cold feeling, or lightheadedness)
- cognitive dysfunction (manifests as alterations of cognitive function or thoughts, including hallucination and illusions)
- emotional dysfunction (manifests as alterations in mood or emotions, including fear, joy, and anxiety)
- autonomic dysfunction (involves the autonomic nervous system and may begin with palpitations, epigastric sensations, pallor/flushing, erection, hypoventilation, urge to urinate or defecate, lacrimation, or pupillary dilation/constriction)
- behavioral arrest (decrease in amplitude or arrest of ongoing motor activity during the seizure; Adamolekun, 2022b; ILAE, 2022e)
Focal seizures can affect a large area of the brain and engage bilateral networks that include both the cortical and subcortical structures, which results in a tonic-clonic seizure and loss of consciousness. These seizures occur when a focal-onset seizure spreads and activates the entire cerebrum. This seizure would be classified as a focal to bilateral tonic-clonic seizure, and the initial focal feature may be brief, making a diagnosis challenging (Adamolekun, 2022b; ILAE, 2022e).
Generalized-Onset Seizures
Generalized-onset seizures originate in both hemispheres; they usually impair awareness and result in a loss of consciousness. These seizures most often result from metabolic disorders and sometimes genetic disorders. Generalized-onset seizures are classified as motor or non-motor, although non-motor seizures may involve some motor activity. With generalized-onset motor seizures, the motor activity is usually bilateral. When the motor activity during a seizure is asymmetrical, differentiating between focal and generalized can be challenging (Adamolekun, 2022b; ILAE, 2022f). Generalized-onset seizures will have the following classifications:
- generalized-onset motor seizures:
- tonic-clonic and variants (formerly known as grand mal)
- tonic (generalized stiffening involving all limbs and without rhythmic jerking)
- clonic (sustained rhythmic jerking)
- atonic (loss of muscle tone)
- myoclonic (rhythmic jerking not preceded by stiffening)
- myoclonic-atonic (myoclonic jerking followed by atonia)
- myoclonic-tonic-clonic (myoclonic jerking followed by tonic and clonic movements)
- epileptic spasms (formerly known as infantile spasms)
- generalized-onset non-motor seizures are further classified based on the first prominent feature:
- typical absence
- atypical absence (less abrupt onset or termination or abnormal changes in tone)
- myoclonic absence
- eyelid myoclonia (Adamolekun, 2022b; ILAE, 2022f)
All absence seizures are classified as general-onset, but distinguishing them from focal impaired awareness seizures can be challenging. However, absence seizures tend to start and end more suddenly and occur more commonly in younger individuals; automatisms are usually less complex than focal impaired awareness seizures. A generalized seizure will have an originating point and rapidly engaging networks distributed bilaterally. They may include cortical and subcortical structures but not necessarily the entire cortex. Each seizure may appear localized, but the location and lateralization are inconsistent across seizures (Adamolekun, 2022b; ILAE, 2022f).
Unknown-Onset Seizures
The third classification, unknown-onset seizure, is used when the seizure has not been categorized as either focal or generalized. Often, this classification is used until more information is available. For example, if a patient enters the emergency department after a seizure, there may be inadequate information to categorize it properly. After a thorough workup, the classification may be updated from unknown onset to either focal or generalized. Seizures with an unknown onset can be motor or non-motor (Adamolekun, 2022b).
Over the years, seizure classification and terminology have evolved (Huff & Murr, 2022). Some seizure types appear in multiple categories within the new classification system. For example, Fisher and colleagues (2017) noted the following changes from 1981 to 2017:
- change in terminology from "partial" to "focal"
- change in seizure types to focal, generalized, or unknown onset
- seizures of unknown onset can have features that are classified
- awareness is a new term used as a classifier of focal seizures
- old terms (i.e., dyscognitive, simple partial, complex partial, psychic, and secondarily generalized) were eliminated from terminology related to seizure activity
- new terms for focal seizure types include automatisms, autonomic, behavior arrest, cognitive, emotional, hyperkinetic, sensory, and focal to bilateral tonic-clonic seizures; atonic, clonic, epileptic spasms, myoclonic, and tonic seizures can be either focal or generalized
- new generalized seizure types include absence with eyelid myoclonia, myoclonic absence, myoclonic-tonic-clonic, myoclonic-atonic, or epileptic spasms (Fisher et al., 2017)
Types of Epilepsy
Epilepsy classification involves three levels: the seizure type, the epilepsy type, and epilepsy syndrome. For clinicians to make the most appropriate diagnosis, a comprehensive history is obtained from a patient and their family. In addition, diagnostic testing is considered, such as imaging and electroencephalography (EEG). The etiology and the classification of epilepsy should be considered and may change as further information is acquired (Adamolekun, 2022b; ILAE, 2022c; Karceski, 2022).
Epilepsy can be classified as follows (ILAE, 2022c):
- Generalized seizures characterize generalized epilepsy and may be accompanied by typical interictal and/or ictal EEG findings. A family history of generalized seizures or epilepsy is supportive of this diagnosis. With genetic generalized epilepsy, generalized epileptiform EEG patterns (i.e., spike wave activity) are known to have a genetic etiology. This classification includes childhood absence epilepsy, juvenile myoclonic and absence epilepsy, or epilepsy with generalized tonic-clonic seizures.
- Focal epilepsy is characterized by focal seizures with typical interictal or ictal EEG findings (i.e., focal sharp waves or interictal slowing). Focal structural brain abnormalities can support this diagnosis. Focal epilepsies can be unifocal, multifocal, or hemispheric.
- Combined generalized and focal epilepsy is characterized by both generalized and focal seizures along with interictal or ictal EEG findings characteristic of each seizure type. Patients with Dravet syndrome or Lennox Gastaut syndrome (LGS) often have this type of epilepsy.
- Unknown epilepsy is used during the diagnostic process to denote epilepsy until a further determination is made between focal, generalized, or combined focal and generalized. This classification can be used when there is insufficient information (e.g. if the EEG is normal or uninformative).
Epilepsy Syndromes
In addition to identifying the etiology, epilepsy can be categorized into epilepsy syndromes based on clinical and electrical characteristics. These syndromes have a typical age of onset, seizure type, and EEG characteristics. Identifying an epilepsy syndrome is important when considering AEDs, as many syndromes can be aggravated by certain AEDs (ILAE, 2022d). Table 2 outlines epilepsy syndromes by age.
Table 2
Epilepsy Syndromes By Age
Age Category | Epilepsy Syndrome |
Neonatal/Infantile |
|
Childhood |
|
Adolescent/Adult |
|
Any Age |
|
(Adamolekun, 2022b; ILAE, 2022d)
Lennox-Gastaut Syndrome
LGS is a severe form of childhood epilepsy characterized by multiple types of seizures, particularly tonic and atonic. LGS is considered an epileptic encephalopathy, meaning frequent seizure activity and abnormal EEG activity degrade cognitive function and cause behavioral difficulties, including hyperactivity, aggression, or agitation. Autism is common. Intellectual and behavioral challenges make daily life difficult for these individuals. The seizures usually begin in preschool but may not be diagnosed for years. LGS seizures are difficult to manage and continue throughout the patient's life. Children may not exhibit all the features of LGS and require close follow-up to ensure an accurate diagnosis. The onset of LGS is typically 1 to 7 years of age (peak at 3 to 5 years); an onset past 10 is rare. Emotional or developmental delays are usually recognized in a patient prior to LGS; boys are diagnosed more often than girls, but no racial differences are noted. Causes of LGS include brain malformations, perinatal asphyxia, central nervous system infection, tuberous sclerosis, severe head injury, and hereditary genetic and degenerative or metabolic disorders. One in 4 people suffering from LGS has no known cause (Adamolekun, 2022b; ILAE, 2022g). The following three features confirm the diagnosis:
- multiple seizure types, starting in childhood but emerging over time
- tonic
- atonic or drop attacks
- atypical absence
- myoclonic and
- generalized tonic-clonic
- focal seizures becoming more common in teens and adults
- characteristic EEG pattern with diffuse or widespread background slowing and slow spike-wave bursts in wakefulness with generalized paroxysmal fast activity found in sleep
- cognitive impairment, behavioral problems, or developmental delay may be present before the onset of seizures; they increase in severity over time with the onset of seizure activity (Adamolekun, 2022b; ILAE, 2022g)
Dravet Syndrome
Dravet syndrome is another rare and drug-resistant type of epilepsy beginning in an otherwise healthy infant's first year of life. The onset is typically a prolonged seizure on one side of the body occurring with a fever, with patients losing control of motor activity in the arm, face, foot, or other body parts. An affected infant is developmentally normal before the onset of seizures, with a decline afterward. By late childhood, they develop a crouched walk; these patients typically walk stooped due to non-physiologic tightening of particular ligaments and muscles. Most cases of Dravet syndrome are due to a severe sodium channel alpha-1 subunit (SCN1A) gene mutation and are associated with varying degrees of developmental disability. In most cases, the mutation is not present in the parents and is considered a new or random mutation in the child (Adamolekun, 2022b; ILAE, 2022a).
Seizures are typically tonic-clonic or clonic on one side of the body. The seizures last longer than 5 minutes and can result in status epilepticus. They recur every few weeks in infancy and early childhood. Seizure types vary among myoclonic, tonic-clonic, atypical, atonic, focal aware or impaired awareness, and tonic seizures. While the initial seizure is most often triggered by fever, subsequent seizures can be triggered by flashing lights, emotional stress, or excitement. An affected child develops appropriately up to age 2 but then misses developmental milestones and progresses slower as they age. A child with Dravet syndrome may have a low motor tone, an unsteady gait, crouched walking as they age, growth or nutritional deficits, and autonomic nervous system dysfunction, which may affect the heart, lungs, stomach, or intestines. This syndrome has mistakenly been linked to vaccines as children develop it near routine vaccinations, but numerous research studies have shown no linkage. Dietary therapy, vagus nerve stimulation, and combination medication therapies are used to control seizures (Adamolekun, 2022b; ILAE, 2022a).
Pathophysiology of Seizures
With advances in modern neuroimaging and genetic testing, the underlying etiologies of epilepsy are more commonly understood and more precisely described (ILAE, 2022b). Epilepsy etiology can include various factors:
- genetic: chromosomal or gene abnormalities
- structural: malformations of cortical development, vascular malformations, hippocampal sclerosis, traumatic brain injury, or tumors
- metabolic: creatinine disorders, folic acid-responsive seizures, glucose transporter 1 (GLUT1) deficiency, or cerebral folate deficiency
- immune: Rasmussen syndrome or antibody-mediated etiologies
- infectious: bacterial meningitis, cerebral malaria, cerebral toxoplasmosis, cytomegalovirus (CMV), HIV, or tuberculosis
- unknown: the underlying cause is not known (ILAE, 2022b)
The pathophysiology of seizures can be quite complex; however, a simplistic explanation is a distortion of the normal balance between excitation and inhibition in the brain. Everyone has some propensity to have a seizure, with a threshold varying along a continuum based on factors that increase susceptibility. The pathophysiology of focal seizures differs from generalized seizures, yet the overall cellular excitability from the imbalance of excitation and inhibition is increased with both forms. The imbalance can occur due to various alterations at many levels of brain function, from genetic or cellular signaling to broad neuronal circuits. The causes of the imbalances can be genetic or acquired. Genetic causes can occur at the circuit and receptor levels or abnormal ionic channel functioning, such as potassium channel mutations. Monogenic or polygenic mutations can result in epilepsy. Acquired cerebral injuries can also alter the circuit function, such as a prolonged fever or head trauma that causes alterations in hippocampal circuitry, leading to seizure activity. The developing brain in an infant or a child is especially prone to seizures with all the physiological changes taking place. However, seizures in children are less likely to cause structural damage than seizures in adults. Since the brain is involved in all bodily functions, multiple body areas can be affected during seizure activity (Huff & Murr, 2022; Ko, 2022).
Seizures start when susceptible cerebral neurons are excited, leading to synchronous discharges of larger groups of connected neurons. Glutamate is the most common excitatory neurotransmitter, while gamma-aminobutyric acid (GABA) is an inhibitory neurotransmitter. Excessive excitation initiates abnormal electrical activity known as electrical paroxysmal depolarization shifts (PDS), which trigger epileptiform activity. The part of the brain affected will often determine the clinical features of the seizure. Generalized convulsive status epilepticus is accompanied by systemic changes that include increased catecholamine levels, hyperthermia, lactic acidosis, and respiratory compromise. In addition, the ongoing excessive electrical activity damages the brain (Huff & Murr, 2022). While the pathophysiology of seizure types may differ for focal onset versus generalized onset, the terms atonic, clonic, myoclonic, and tonic are used for both categories (Fisher et al., 2017).
Precipitating Factors
The triggers of seizures and epilepsy are numerous. They can vary based on the type of seizure (i.e., focal or generalized) and the seizure setting (i.e., acute symptomatic [occurs at the time of a systemic or brain insult] versus unprovoked). Acute symptomatic seizures are triggered by precipitating factors that are environmental, physical, clinical, or emotional. These triggers may vary from patient to patient. However, common themes are physical or emotional stress, alcohol or drug use, illness (particularly with increased temperatures or hypoglycemia or hyperglycemia), menstruation, or sleep deprivation. Missing doses of antiepileptic drugs (AED) or taking medications that interfere with the function of AEDs can also trigger a seizure. Further triggers in those with epilepsy include strobe lights or video games. Patients and caregivers/parents should be counseled to maintain a record or journal of seizure triggers, as these can help lessen the likelihood of a seizure being triggered in the future (Huff & Murr, 2022; Schachter, 2022a). Other common causes of seizures can include:
- acute toxic effects (e.g., sympathomimetics, antidepressants)
- electrolyte disturbances (e.g., hyponatremia, hypernatremia, hypocalcemia, hypomagnesia)
- withdrawal syndrome (e.g., benzodiazepines)
- sepsis
- CNS infection
- hypoxic brain injury
- ischemic or hemorrhagic stroke
- neoplasm
- traumatic brain injury
- inflammatory (e.g., receptor encephalitis, lupus cerebritis)
- hyperthyroidism
- uremia
- eclampsia (Huff & Murr, 2022; Schachter, 2022a)
Assessment and Diagnosis
Diagnosing seizures can be a challenge for healthcare providers (HCPs). Many disorders can cause behavioral changes, which can be confused with seizures or epilepsy. As the treatment of seizures requires an accurate diagnosis, determining the type of seizure or epilepsy is vital. HCPs must obtain clear descriptions of seizure activity, including the timing, potential triggers, symptoms, longevity, or any other descriptors that can aid in diagnosis. Patients with a new-onset seizure are typically evaluated within the emergency department and are often discharged after a thorough evaluation. Evaluation can often occur in the primary HCP office for patients with a known seizure disorder. Then, they may be referred to a neurologist or epilepsy specialist known as an epileptologist. An epileptologist is often used for high-risk cases such as pregnancy and childbirth or uncontrolled seizures. Identifying any underlying cause for the seizures and the location within the brain where the seizures are being generated is crucial. A detailed medical history, blood tests, EEG, and brain imaging—including a computer tomography (CT) scan, magnetic resonance imaging (MRI), or positron emission tomography (PET) scans—are needed to diagnose the underlying etiology, specifically new-onset seizures. The diagnostic workup for a patient with a known seizure disorder may be limited to assessing AED levels (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a).
Epilepsy should only be diagnosed after ruling out epilepsy imitators, including the following:
- syncope and anoxic seizures (i.e., non-epileptic events caused by temporary impairment of blood flow to the brain)
- behavioral, psychological, or psychiatric disorders
- sleep-related conditions
- paroxysmal movement disorders
- migraine-associated disorders
- miscellaneous events, including:
- shuddering attacks (i.e., non-epileptic stiffening, tonic posturing, or rapid shivering movements)
- jitteriness
- spasmus nutans (i.e., a rare, idiopathic disorder that involves nystagmus, head nodding, and torticollis)
- increased intracranial pressure
- spinal myoclonus
- paroxysmal extreme pain disorder (ILAE, 2022e)
Medical History
A detailed history will be taken during the initial visit. This aims to characterize the seizure, rule out alternative diagnoses, and evaluate for underlying risk factors, including medical and family history and medications (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a). The history should include questions such as:
- Can you give me a description of what happened?
- Is there a family member or witness to the seizure or event who can give further descriptions, including information on loss of consciousness?
- What events led up to the event or seizure?
- Was there any drug or alcohol use prior to the event or seizure?
- Were you sleep-deprived leading up to the event or seizure?
- What was the setting of the event or seizure?
- Did you just stand up or change positions before the event or seizure?
- Was there a warning?
- How long did it last?
- Were there any after-effects following the event or seizure?
- Were you tired or confused after the event or seizure?
- Has there been more than one event or seizure?
- Have you ever seen an HCP for this condition before?
- If so, what tests have been run, and when?
- Have you been prescribed any medication for the condition? If so, what? What effect did it have? (Epilepsy Foundation, 2013)
An accurate description of the seizure is essential but can be challenging to obtain. Patients with focal seizures without impairment can usually describe the event, while those with focal seizures with impairment or generalized seizures cannot. Most seizures have a clear and abrupt clinical onset and rapid progression of symptoms over seconds. In addition, most seizures end spontaneously within 2 to 3 minutes. Ictal behaviors help determine the origin of a seizure and whether it is focal or generalized. See Table 3 for clinical manifestations of focal seizures by brain location. After the seizure, most patients will exhibit postictal confusion and decreased alertness. Some patients may also experience focal neurological deficits, including aphasia, weakness, or numbness. The postictal state can last seconds to several days, depending on the seizure type and underlying brain function. Most patients begin to recover alertness and responsiveness within 10 to 20 minutes of a generalized seizure, and prolonged symptoms warrant further evaluation (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a). HCPs should use the questions above to assess for precipitants or triggers for the seizure.
Table 3
Clinical Manifestations of Focal-Onset Seizures by Brain Location
Clinical Manifestation | Brain Location of Dysfunction |
Simple movements (e.g., limb twitching) | Contralateral frontal lobe |
Abnormal taste | Insula |
Olfactory hallucinations | Anteromedial temportal lobe |
Complex behavioral automatisms | Temporal lobe |
Visual hallucinations | Occipital lobe |
Bilateral tonic posture | Frontal lobe |
Head and eye deviation with posturing | Supplementary motor cortex |
Visceral or autonomic abnormalities (e.g., salivation or epigastric aura) | Insular-orbital-frontal cortex |
Chewing movements, speech arrest, salivation | Amygdala, opercular region |
Unusual behavior suggesting a sleep disorder or psychiatric cause | Frontal lobe |
Localized sensory disturbances (e.g., numbness or tingling of limb or half the body) | Parietal lobe |
(Adamolekun, 2022b)
Physical Examination
HCPs should conduct a physical examination for any patients who had a seizure. In addition to a general physical examination, a focused neurological examination is critical to assess for any deficits. For patients who have lost consciousness, HCPs should look for incontinence, a bitten tongue, or prolonged confusion to suggest a seizure. Posterior shoulder dislocation can occur during a seizure; therefore, any new shoulder pain or limited range of motion should warrant radiographic examination. HCPs should also look for signs of psychogenic non-epileptic seizures (PNES), previously known as pseudoseizures, which are paroxysmal changes in behavior that resemble epileptic seizures. However, PNES lacks an organic cause, and the expected EEG findings present with epileptic seizures (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a). PNES can resemble tonic-clonic generalized seizures but have distinguishing clinical characteristics:
- postictal confusion tends to be absent
- they last longer (several minutes or more)
- typical seizure activity usually does not occur (tonic phase, followed by clonic phase)
- muscular activity usually includes jerking from one side to the other and back and exaggerated pelvic thrusting
- vital signs remain normal
- patients often resist passive eye-opening
- intensity waxes and wanes (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a)
Diagnostic Testing
Since patients may have other diagnoses besides seizures, further testing may be needed. However, normal results do not necessarily exclude a seizure; therefore, the diagnosis may be clinically based. The gold standard for diagnosing a seizure is video EEG monitoring. Particularly for focal impaired awareness seizures or absence status epilepticus, EEG may provide the most definitive indication of a seizure. Additional tests may be ordered, including an MRI of the brain or a CT angiogram of the head and neck, carotid ultrasound, conventional angiography, PET scan, or cardiac Holter monitor. These tests should be used to rule out or confirm other diagnoses, such as transient ischemic attacks (TIAs), stroke, vasculitis, vascular malformations, or cardiac arrhythmias. An overnight sleep study (polysomnography) should be ordered to rule out parasomnias (abnormal movements during sleep) or obstructive sleep apnea. A movement disorder physiologic study may be needed to identify myoclonus or tremor (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a).
Blood tests should be performed to evaluate for infection, blood glucose abnormalities, electrolyte imbalances, blood urea nitrogen (BUN), creatinine, liver function, or genetic conditions. A lumbar puncture should be performed if an infection could be the source of the seizure. In addition, a neurological examination should be completed to assess behavior, mental, and motor functioning to identify any other deficits related to the nervous system (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a).
During an EEG, electrodes are attached to the patient's scalp using a paste-like substance. The electrodes record the electrical activity (impulses) of the brain. With epilepsy, patients often have changes in normal brain activity, as evidenced by alterations in the wave patterns on the EEG results, even when a seizure is not occurring. A video EEG is the preferred diagnostic method as it can provide insight into the patient's response during the seizure; it involves a simple camera recording of the patient during the EEG. Inpatient combined video-EEG monitoring can be done for 2 to 7 days, recording EEG activity and clinical behavior simultaneously. It is possible to perform ambulatory EEGs (without video) in which the patient wears the EEG at home; this provides recordings of the patient's brain activity over several days. A high-density EEG may be ordered to give a more precise determination of the areas of the brain affected by seizures. This EEG requires the electrodes to be much closer together than in a regular EEG, typically 0.5 cm apart. There is no specific patient preparation for an EEG unless sleep deprivation is utilized to help trigger a seizure (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a).
A normal EEG cannot exclude the diagnosis of epileptic seizures, especially when seizures are infrequent. The initial EEG may only detect abnormalities in 30% to 50% of patients with a known epileptic disorder. Serial EEGs can detect abnormalities in up to 80% to 90% of patients (Adamolekun, 2022b). EEG findings may include the following:
- epileptiform abnormalities in the temporal lobe foci between seizures for focal impaired-awareness seizures in the temporal lobe
- focal epileptiform discharges in focal-to-bilateral tonic-clonic seizures
- interictal bilateral symmetric bursts of 4- to 7-Hz epileptiform activity in generalized tonic-clonic seizures
- spikes and slow-wave discharges occurring bilaterally at a rate of 3/second and usually normal background EEG activity in typical absence seizures
- slow spike and wave discharges, usually at a rate of <2.5/second, with interictal disorganization of background activity and diffuse slow waves in typical absence seizures
- bilateral polyspike and wave abnormality at a rate of 4- to 6-Hz in juvenile myoclonic epilepsy (Adamolekun, 2022b)
A CT scan can be used to obtain cross-sectional images of the brain to evaluate for structural abnormalities that may be causing the seizures, such as tumors, cysts, or bleeding. A CT scan is a 3-dimensional (3D) computer-generated graphic based on x-ray images. Similarly, an MRI can detect lesions or abnormalities in the brain using powerful magnets and radio waves. A more in-depth MRI called a functional MRI (fMRI) can measure the changes in blood flow within the brain, as seen in Figure 1a. An fMRI may be used before surgery to locate critical functions precisely, such as movement or speech, so the surgeon can avoid injury to those areas during surgery. PET scans can detect brain function abnormalities. A small amount of radioactive tracer material is typically injected to help visualize chemical activity in certain tissues, as seen in Figure 1b. If the MRI, EEG, or other tests could not determine exactly where the seizures originated, a single-photon emission CT (SPECT) scan might be conducted. This test uses a radioactive tracer to provide a detailed, 3D map of the blood flow through the brain during seizures (Adamolekun, 2022b; Haider & Bullinger, 2022).
Figure 1
fMRI (a) and PET (b) Brain Scan Images
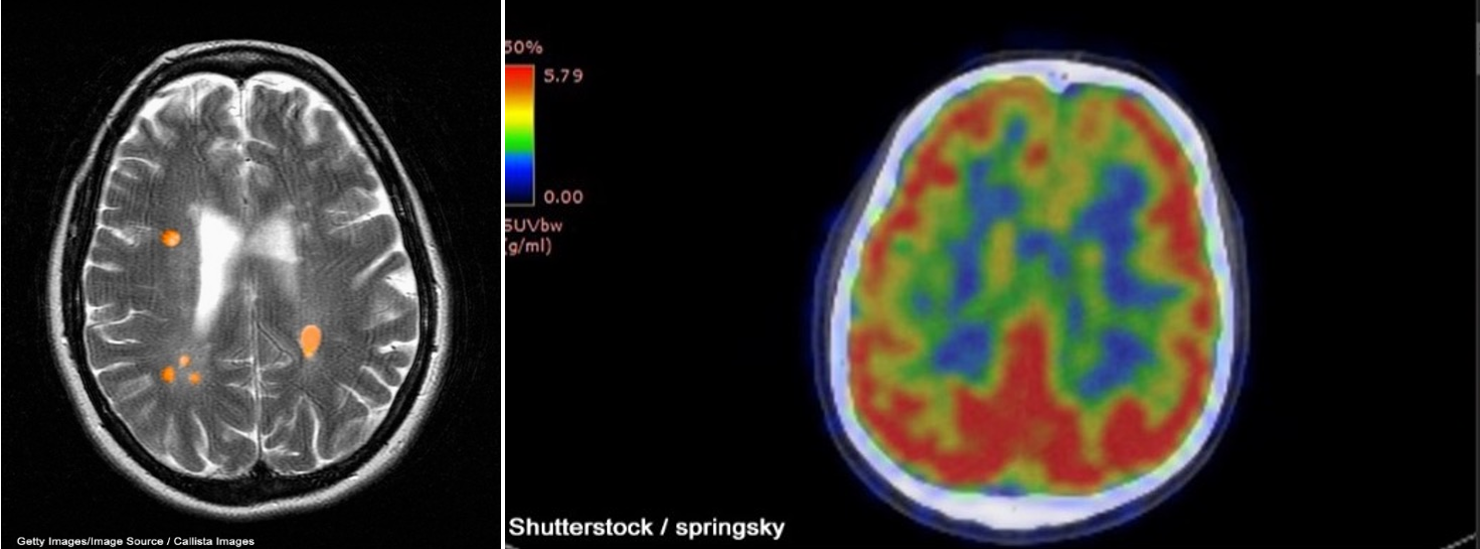
In addition to diagnostic testing, statistical parametric mapping (SPM), Curry analysis, or magnetoencephalography (MEG) can help determine exactly where the seizures originated. SPM is a method where tissue metabolism is assessed, as brain areas have an increased metabolic rate during seizures, to determine where the seizure originated. Curry analysis combines EEG data and an MRI of the brain to determine where the seizures are occurring. MEG measures the magnetic fields created through electrical brain activity to locate seizure onset areas (Haider & Bullinger, 2022).
Once an accurate diagnosis is established by identifying the seizure type and location of origin, proper treatment can be initiated. Some people may have a single seizure that is determined to be associated with an underlying condition; once this condition has been treated, the patient should not have any further seizures. However, ongoing seizures may be diagnosed as epilepsy (Haider & Bullinger, 2022).
Management of Epilepsy/Seizures
Acute Management
Since seizures have multiple etiologies not associated with epilepsy, the treatment modalities will also vary. Treatment for seizures not due to epilepsy will focus on finding the underlying condition and correcting it while also halting the seizure activity. Most seizures will resolve spontaneously within 2 minutes and do not require rapid administration of an AED. However, HCPs or prehospital personnel should secure intravenous (IV) access in case of subsequent seizures. If a subsequent seizure occurs or prolonged seizure activity is present, IV medications may be administered. In addition, if the cause of the seizure cannot be identified and corrected, medication should be considered. Medications are often unnecessary if the underlying cause is identified and treated since the risk of a subsequent seizure is low. Typical IV medications for seizures include benzodiazepines, such as lorazepam (Ativan) and diazepam (Valium). A patient diagnosed with epilepsy may be given IV benzodiazepines to stop a seizure, but repeated oral doses of a benzodiazepine are typically unnecessary. Oxygen therapy should be used as indicated. Patients having a seizure should never be restrained. In addition, nothing should be put in their mouth that could cause further injury, such as a bite guard (Adamolekun, 2022b; Huff & Murr, 2022; Schachter, 2022a).
AEDs are required to stop the seizure activity for patients with seizures lasting longer than 5 minutes or those in status epilepticus. The sooner these medications are initiated, the quicker the seizure activity can be controlled. These patients should be given lorazepam (Ativan) 0.05 to 0.1 mg/kg IV (typically 4 mg for adults) at a maximum of 2 mg/minute. Some patients may require a larger dose. If IV access is unavailable, midazolam (Versed) can be administered IM or diazepam (Valium) can be given intranasally or rectally. After lorazepam (Ativan) is given, a second, longer-acting AED should be given. No consensus exists on which longer-acting medication should be given (Adamolekun, 2022b; Drislane, 2022). The most common options include the following:
- fosphenytoin (Cerebyx) 15 to 20 phenytoin equivalents (Pes)/kg IV, given at a rate of 100 to 150 PE/minute (fosphenytoin [Cerebyx] is given in PEs, with fosphenytoin [Cerebyx] 1.5 mg being equivalent to phenytoin [Dilantin] 1 mg)
- levetiracetam (Keppra) loading dose of 60 mg/kg (max 4500 mg) infused over at least 15 minutes, followed by 1500 mg orally BID
- valproate sodium (Depacon) 20 to 40 mg/kg IV loading dose (maximum 3000 mg) at 10 mg/kg per minute, followed by 4 to 8 mg/kg orally TID
- phenytoin (Dilantin) 15 to 20 mg/g IV, at a rate of 50 mg/minute; this cannot be given through the same IV line as a benzodiazepine or any fluid with glucose/dextrose (Adamolekun, 2022b; Drislane, 2022)
If seizures persist after one of these options, an additional dose of fosphenytoin (Cerebyx) 5 to 10 PE/kg or phenytoin (Dilantin) 5 to 10 mg/kg should be given. If a third AED is warranted due to persistent seizure activity, phenobarbital (Luminal), propofol (Diprivan), midazolam (Versed), levetiracetam (Keppra), and valproate (Depacon) are recommended. Phenobarbital (Luminal) is given at a dose of 15 to 20 mg/kg IV at a rate of 100 mg/minute, with a second dose of 5 to 10 mg/kg if seizures persist. If status epilepticus is not resolved after a third AED, intubation and general anesthesia are necessary (Adamolekun, 2022b).
Similarly, rescue treatments may be needed during seizure activities outside a hospital or clinical setting. Many patients have emergency medications to be administered by others during an acute seizure (not routine medications that are taken daily), such as diazepam (Valium) or other benzodiazepines, to stop the seizure quickly. These drugs can be prescribed for emergency use with clear instructions on administration by family or friends outside a clinical setting. Emergency care may still be needed, depending on the situation. Rescue medications may be used for seizures that are unusual or last longer than typical events, cluster seizures (multiple short seizures), or breakthrough seizures that may occur during medication changes or with an acute illness. Diazepam (Valium) can be given rectally, or a dissolvable tablet can be placed under the tongue or between the cheek and gum for quick absorption. Benzodiazepines are not generally recommended for daily use due to their risk of abuse, dependence, or increased fall risk, particularly for adults over 65 (Adamolekun, 2022b).
The most important consideration is safety during seizures. Caregivers should be instructed to provide a safe environment during a seizure. Patients should be gently helped to the floor and placed on their side, and their airway must be kept clear. Clothing around the neck should be loosened, and something soft, such as a pillow, should be placed under the head. The caregiver should stay with the patient and observe any behaviors that can be reported to the HCP later to support the diagnosis and treatment plan. Caregivers should call 911 if the seizure lasts more than 5 minutes, the patient does not return to their typical mental and physical state, the patient is injured or pregnant, there are repeated seizures, the seizure is the first for the patient, the patient has difficulty breathing, or the seizure occurs in water (Adamolekun, 2022b).
Maintenance Treatment: Pharmacological
Epilepsy patients are typically placed on long-term AED therapy. However, no single drug can control all types of seizures, and many patients require different drugs. Roughly 60% of seizures are controlled by a single AED, while 30%-40% require a combination. The preferred AED varies based on seizure type, potential adverse effects of the drug, interactions with other medications, cost, patient age, childbearing plans, and comorbid conditions. HCPs should consider that some AEDs reach therapeutic ranges quickly while others increase gradually over weeks. In addition, patient tolerance for AEDs can vary significantly; therefore, dosing should start low and gradually increase while monitoring drug toxicity. The goal of AED therapy should be the lowest dose needed to stop all seizures with the fewest side effects. The use of multiple AEDs should be avoided if possible due to poor adherence and the risk of drug interactions, with the incidence of adverse effects doubling. Once seizures are controlled, the patient should continue therapy until seizure-free for at least 2 years. HCPs can then consider weaning the AED using a slow taper (i.e., 10% every 2 weeks). If the patient relapses, AEDs should be restarted and continued indefinitely. Table 4 reviews the choice of drugs based on the seizure type, and Table 5 reviews the most common AEDs and their associated nursing implications. AEDs are typically grouped by mode of action, although the precise mechanism of action is not known for many, and others have multiple actions (Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023).
Table 4
Choice of Antileptic Drug Based on Seizure Type
Seizure Type | AEDs | Use |
Generalized-onset tonic-clonic seizures | Divalproex (Depakote ER) Valproic acid (Depakene) | First-line monotherapy |
Lamotrigine (Lamictal) Levetiracetam (Keppra) Topiramate (Topamax) | Second-line monotherapy or adjunctive therapy | |
Perampanel (Fycompa) Zonisamide (Zonegran) | Adjunctive therapy | |
Phenobarbital (Luminal) | Second-line monotherapy due to sedating effects and possible behavioral problems in children | |
Various types of focal-onset and focal-to-bilateral tonic-clonic seizures | Carbamazepine (Tegretol) Fosphenytoin (Cerebyx) Lamotrigine (Lamictal) Levetiracetam (Keppra) Oxcarbazepine (Trileptal) Phenytoin (Dilantin) Topiramate (Topamax) | First-line monotherapy |
Cenobamate (Xcopri) Divalproex (Depakote ER) Eslicarbazepine (Aptiom) Gabapentin (Neurontin) Lacosamide (Vimpat) Perampanel (Fycompa) Pregabalin (Lyrica) Valproic acid (Depakene) Zonisamide (Zonegran) | Second-line monotherapy or adjunctive therapy | |
Clobazam (Frisium) Felbamate (Felbatol) Tiagabine (Gabitril) Vigabatrin (Sabril) | Third-line monotherapy or adjunctive therapy | |
Phenobarbital (Luminal) | Effective but less desirable due to sedating effects and can cause behavioral problems in children | |
Typical non-motor absence seizures | Divalproex (Depakote ER) Ethosuximide (Zarontin) Lamotrigine (Lamictal) Valproic acid (Depakene) | First-line monotherapy |
Clobazam (Frisium) Levetiracetam (Keppra) Topiramate (Topamax) Zonisamide (Zonegran) | Also effective | |
Atypical non-motor absence seizures Non-motor seizures associated with other seizure types | Divalproex (Depakote ER) Felbamate (Felbatol) Lamotrigine (Lamictal) Topiramate (Topamax) Valproic acid (Depakene) | First-line monotherapy |
Clonazepam (Klonopin) | Effective but often develops tolerance | |
Acetazolamide (Diamox) | Reserved for refractory cases | |
Epileptic (infantile) spasms Atonic seizures Myoclonic seizures | Divalproex (Depakote ER) Valproic acid (Depakene) | First-line monotherapy |
Vigabatrin (Sabril) | Risk of irreversible visual field defects | |
Clonazepam (Klonopin) | Second-line therapy | |
Dravet syndrome | Cannabidiol (Epidiolex) Clobazam (Frisium) Topiramate (Topamax) Valproic acid (Depakene) | Adjunctive therapy |
Tonic and/or atonic seizures in LGS | Divalproex (Depakote ER) Lamotrigine (Lamictal) Topiramate (Topamax) Valproic acid (Depakene) | First-line monotherapy |
Cannabidiol (Epidiolex) | Adjunctive therapy | |
Clobazam (Frisium) Felbamate (Felbatol) Zonisamide (Zonegran) | Alternative or adjunctive therapy for atonic seizures | |
Divalproex (Depakote ER) Valproic acid (Depakene) | First-line monotherapy | |
Lamotrigine (Lamictal) Levetiracetam (Keppra) Topiramate (Topamax) Zonisamide (Zonegran) | Second-line monotherapy or adjunctive therapy | |
Unclassified seizures | Divalproex (Depakote ER) Valproic acid (Depakene) | First-line monotherapy |
Lamotrigine (Lamictal) | Second-line monotherapy | |
Levetiracetam (Keppra) Topiramate (Topamax) Zonisamide (Zonegran) | Third-line monotherapy or adjunctive therapy |
(Adamolekun, 2022a, 2022b)
Table 5
Common Antiepileptic Drugs
Medication: diazepam (Diastat, Intensol, Valium) Dosing:
Nursing Implications/Patient Education:
|
Medication: lorazepam (Ativan) Dosing:
Nursing Implications/Patient Education:
|
Medication: phenytoin (Dilantin) Dosing:
Nursing Implications/Patient Education:
|
Medication: phenobarbital (Luminol) Dosing:
Nursing Implications/Patient Education:
|
Medication: valproic acid (Depakene; oral), valproate sodium (Depacon; intravenous), divalproex sodium (Depakote DR/ER, sprinkles; oral) Dosing:
Nursing Implications/Patient Education:
|
Medication: topiramate (Topamax) Dosing:
Nursing Implications/Patient Education:
|
Medication: carbamazepine (Tegretol) Dosing:
Nursing Implications/Patient Education:
|
Medication: clonazepam (Klonopin) Dosing:
Nursing Implications/Patient Education:
|
(Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023; Tucker, 2022)
HCPs should educate patients to take AEDs at the same time every day and maintain a routine, either with or without food, as appropriate. Consistency is vital to maintaining a therapeutic blood level and controlling seizure activity. Patients should be warned never to stop their medication(s) without the prescriber's knowledge and direction. Most medications must be tapered to avoid withdrawal symptoms or increased seizure activity (Schachter, 2022b).
Cannabis use in the treatment of epilepsy has also been studied. The medicinal properties of cannabis have been credited mainly to the two most biologically active phytocannabinoids, Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD; Mouhamed et al., 2018). CBD was isolated in 1940, and THC was isolated in 1964. In the 1990s, the human endocannabinoid system was discovered, and the cannabinoid receptors—CB1 and CB2—were cloned. In 2015, Rosenberg and colleagues evaluated four primary clinical trials to examine the efficacy and safety of medical cannabis (CBD) in seizures. Two studies showed a partial antiseizure effect with CBD, and two showed no statistically significant effect. However, these studies were small, with insufficient blinding or randomization and incomplete datasets (Rosenberg et al., 2015). Mouhamed and colleagues (2018) assessed treatment with THC as too broad for therapeutic purposes. Still, they found positive findings when they reviewed five trials published between 2013 and 2018 studying the use of CBD in treating drug-resistant epilepsy among children or young adults. These studies showed a more significant reduction in the frequency of atonic and partial seizures, followed by reductions in tonic/tonic-clonic seizures, with minimal reports of adverse effects. Of note, these studies did not utilize CBD as a replacement for the subjects' AED but as an adjunct. In 2018, the FDA approved cannabidiol (Epidiolex) for treating seizures associated with LGS and Dravet syndrome in patients 2 years and older. This is the first FDA-approved drug that contains a purified substance derived from marijuana. The efficacy of cannabidiol (Epidiolex) was evaluated through 3 randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either LGS or Dravet syndrome. Within these studies, when cannabidiol (Epidiolex) was taken alongside other medications, it reduced the frequency of seizures better than placebo. The most common side effects include sedation, sleepiness, fatigue, weakness, malaise, and lethargy. Less common side effects include elevated liver enzymes, decreased appetite, diarrhea, skin rash, and insomnia. Cannabidiol (Epidiolex) is an oral solution of 100 mg/mL. For LGS and Dravet syndrome, the initial dose is 2.5 mg/kg orally BID that can be increased after 1 week to the suggested maintenance dose of 5 mg/kg BID. The maximum dose is 10 mg/kg BID (20 mg/kg daily). Seizures/epilepsy is one of the most common qualifying medical conditions in the US for legalized medical cannabis/marijuana (FDA, 2020; Schachter, 2023).
Nonpharmacological and Self-Management Strategies
Epilepsy can impact an individual in various ways. HCPs must tailor the treatment plan to the individual's unique needs. A new seizure diagnosis can accompany a loss of independence, employment, ability to drive, and self-esteem. Therefore, HCPs should address psychosocial issues when creating a plan of care (Adamolekun, 2022a; Epilepsy Foundation, 2019). Patients and their families should be given tools to manage this disorder, including:
- a medication list and schedule
- a healthcare contact list with phone numbers and other contact information for all HCPs and local EMS
- an online seizure diary or physical journal that maintains information about seizure activity and potential triggers
- a dietary diary (Adamolekun, 2022a; Epilepsy Foundation, 2019)
Empowering patients with tools to prepare them for adverse events, prevent future events, and ensure preparedness in all situations is important. The Epilepsy Foundation (2019b) offers an online diary and a mobile application to document details about an individual's seizure experience. The online resource is called My Seizure Diary. This self-management tool and medication list allows patients to track their health history, manage medication and other therapies, recognize triggers through tracking, and even communicate with their HCP. Seizure alert devices are also available; they help alert family members that a seizure is occurring. This can be particularly helpful for children to alert caregivers or family members of impending seizure activity. Service animals can also be trained to alert patients of impending seizure activity or alert caregivers when a seizure occurs, maintaining protection of the patient as well as activating an alert for help (Adamolekun, 2022a; Epilepsy Foundation, 2019).
Dietary therapy can be beneficial in the management of epilepsy. Diets like the classic ketogenic diet ( a high-fat, low-carbohydrate, moderate-protein diet) can help control seizures in many patients. These diets are used in conjunction with medications. The ketogenic diet focuses on the ratio of fat to carbohydrate and protein grams combined. Carbohydrates and proteins are restricted, and typically there is a 3:1 or 4:1 ratio of fat to carbohydrates/proteins. An HCP or a dietician can prescribe a specific number of calories, and the calories will be focused on foods high in fat, such as heavy whipping cream, butter, mayonnaise, and olive or canola oil. This diet is usually started in the hospital with extensive dietary teaching for the family and may begin with an 18- to 24-hour fast except for water to expedite ketosis. The patient and/or their family must track the carbohydrates in all foods and medications. Children who follow this diet may decrease their seizures by up to 50%, and another 10% to 15% become seizure-free on this diet. Most continue to take their medications; however, the diet may allow them to decrease the dose required. Side effects of this diet include kidney stones, high cholesterol, constipation, slowed growth, and bone fractures. Dietary supplements are typically needed, as this diet does not provide sufficient amounts of all essential micronutrients (e.g., calcium, vitamin D, and selenium). Other diets that have shown success are the low glycemic index diet, the medium-chain triglyceride (MCT) ketogenic diet, and the modified Atkins diet (Epilepsy Foundation, 2017).
Other treatment options that should be considered when medication and diet do not control seizures or cause intolerable side effects include surgery or implantable devices. If seizures are intractable, the patient should be referred to an epilepsy center to be evaluated for surgery. Surgeons may elect to remove the part of the brain that is causing seizures. Neuromodulation is electrical stimulation using a small implanted device that sends electrical currents to the affected area of the brain. Various devices are available, and each works differently; generally, the stimulation releases substances that change how the cells act and restores their normal state (Adamolekun, 2022a).
Pregnancy Considerations
Pregnancy can bring unique challenges to managing epilepsy. Women are affected by epilepsy differently than men, partially due to hormonal changes, menstrual cycles, and menopause. Estrogen increases the brain's electrical activity, and progesterone has the opposite effect. AEDs are often adjusted due to the different life phases of women, and pregnancy poses challenges to managing medications and protecting fetuses. AEDs are associated with an increased risk of teratogenicity, such as fetal antiepileptic drug syndrome (i.e., cleft lip, microcephaly, developmental delay, cardiac defects, growth restriction, and abnormal facies, limbs, or digits). Over 90% of women with a history of epilepsy have healthy babies, yet 10% may have congenital disabilities. The risk of seizure activity during pregnancy increases by 25%, and many require medication adjustments. In most cases, women with a history of epilepsy do not have difficulty becoming pregnant. However, valproate (Depakene, Depakote) can contribute to polycystic ovarian syndrome, making conception more difficult (Adamolekun, 2022a; Schachter, 2022b).
When considering pregnancy, women with epilepsy should plan ahead, as planned pregnancies have opportunities for the greatest outcomes. Seizures can cause falls, auto accidents, premature labor, miscarriage, or decreased oxygen delivery and heart rate in the baby. The healthcare team for any patient considering pregnancy should include a neurologist or an epilepsy specialist and an obstetrician to develop a treatment plan ahead of time that provides optimal opportunities for the mother and baby. Unfortunately, approximately 50% of all pregnancies in the US are unplanned. A pre-pregnancy plan should include a thorough review of AEDs. Baseline pre-pregnancy AED drug levels should be obtained, and ongoing pregnancy monitoring should occur as hormonal changes cause fluctuating medication levels. Many AEDs decrease B12 and folate levels; therefore, preconception supplements are needed to support nutritional deficiencies and decrease the risk of neural tube defects associated with some medications, particularly in the first trimester. The suggested daily dose of folic acid is a multivitamin with 400 mcg folate and daily supplementation of up to 4 mg/day for 1 to 2 months prior to pregnancy for women taking valproate (Depakene, Depakote) or carbamazepine (Tegretol). Patients hoping to become pregnant should pay close attention to seizure triggers and ensure healthy living with a healthy diet and exercise. There are no reported increases in seizures during delivery; however, if a seizure occurs, medications should be given to control it immediately. A Caesarean section should be considered if the mother is at risk of seizure or experiences a prolonged seizure during labor (Adamolekun, 2022a; Schachter, 2022b).
Medications to prevent seizures must be continued during pregnancy, as harm can come to the baby and mother with seizure activity. Risks increase when the mother takes multiple AEDs and at higher doses. Due to these risks to the fetus, valproate (Depakene, Depakote) should be avoided during pregnancy if possible. Phenobarbital (Luminal), pregabalin (Lyrica), and topiramate (Topamax) pose an intermediate risk; specifically, topiramate (Topamax) increases the risk of a cleft palate if used in early pregnancy. The lowest-risk AEDs are carbamazepine (Tegretol), lamotrigine (Lamictal), and levetiracetam (Keppra). At this time, there are many unknowns about AED use in pregnancy, but it is undeniable that the risk increases with higher dosages and multiple medications. Pregnant mothers should take the minimum effective dose of a single medication to control seizures whenever possible. Regarding breastfeeding, most studies monitoring children up to 6 years of age born to mothers taking AEDs found they had equal or higher IQs when compared to children who were bottle-fed. Therefore, mothers are advised to take their AED immediately after breastfeeding to decrease the amount of medication in the breast milk (Adamolekun, 2022a; Schachter, 2022b).
Older Adult Considerations
Seizure rates are highest in the first few years of life, taper through the 50s, and rise again later in life. Seizures increase with other age-related conditions like Parkinson's disease but also rise with renal failure, electrolyte or glucose disturbances, drug intoxication or withdrawal, trauma, degenerative illnesses like Alzheimer's disease, infectious diseases such as meningitis or encephalitis, and strokes. Treating adults over 65 can be challenging due to their sensitivity and reactions to AEDs. Gabapentin (Neurontin) and lamotrigine (Lamictal) have advantages for older adults due to few interactions with other drugs. Gabapentin (Neurontin) is approved as a single therapy for seizures with high efficacy. Generally, carbamazepine (Tegretol) or phenytoin (Dilantin) may decrease the effectiveness of other drugs that older adults are often taking, such as warfarin (Coumadin). If warfarin (Coumadin) is taken with any of these medications, the prothrombin time (PT) must be checked frequently, and the dose must be adjusted accordingly. These medications also lower folate and vitamin D levels, which are important in managing osteoporosis and decreasing fractures in older women. The four primary enzyme-inducing AEDs are carbamazepine (Tegretol), phenytoin (Dilantin), phenobarbital (Luminol), and primidone (Mysoline; Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023). These medications stimulate metabolism and decrease the serum concentration of other AEDs, as well as:
- warfarin (Coumadin)
- acetaminophen (Tylenol)
- digoxin (Lanoxin)
- furosemide (Lasix)
- propranolol (Inderal)
- quinidine (Cardioquin)
- theophylline (Theo-Dur)
- tricyclic antidepressants such as amitriptyline (Elavil; Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023)
These AEDs also inhibit the metabolism of the following medications, often requiring dosage adjustments to avoid toxicity:
- amiodarone (Pacerone)
- cimetidine (Tagamet)
- ranitidine (Zantac)
- erythromycin (Erythrocin)
- nifedipine (Procardia XL)
- isoniazid (Niazid)
- verapamil (Calan)
- trimethoprim (Primsol; Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023)
Gabapentin (Neurontin) and lamotrigine (Lamictal) are not metabolized in the same way and do not cause these interactions. Other potential adverse effects of AEDs for older adults include:
- increased risk of falls due to ataxia from the medications
- risk of bradycardia or other arrhythmias from carbamazepine (Tegretol)
- risk of hyponatremia with carbamazepine (Tegretol) or oxycarbazepine (Trileptal)
- thrombocytopenia, parkinsonism, dementia, and hearing deficits with valproate (Depakene, Depakote; Adamolekun, 2022a, 2022b; Schachter, 2022b, 2023)
Beer's Criteria are guidelines used to direct appropriate medications for older adults. The criteria were published in 1991 and were intended to improve the selection of medications for those 65 and older. The criteria are updated periodically. The latest update lists benzodiazepines as potentially inappropriate medications (PIMs); they should be cautiously administered to older adults due to their increased sensitivity and slower ability to metabolize these medications. There is also a risk of impaired cognitive functioning, delirium, falls, fractures, and accidents in and out of the home with these drugs (Fick et al., 2019; Schachter, 2022b).
Febrile Seizures in Children
A febrile seizure is a common seizure disorder in pediatric patients associated with fever without other intracranial abnormalities or infections. Febrile seizures can be classified into simple, complex, and symptomatic. Children with a genetic predisposition can be more susceptible to febrile seizures. Viruses, bacteria, or vaccines can be causative agents of febrile seizures. Simple febrile seizures last under 15 minutes, are generalized, occur once within 24 hours, and have no focal component. A complex febrile seizure lasts longer than 15 minutes, has a focal component, or occurs more than once in 24 hours. A symptomatic febrile seizure is due to a fever in a child with a preexisting neurological abnormality. Acute management of a febrile seizure is to maintain a patent airway, ensure effective breathing, provide oxygen therapy as needed, protect the patient from injury, place them in a semi-prone position and preferably on the right side, loosen their clothing, and treat the fever with antipyretics such as acetaminophen (Tylenol) or ibuprofen (Motrin). According to the duration and severity, rectal diazepam can be given for any seizure over 5 minutes in length, or IV diazepam (Valium), lorazepam (Ativan), or phenobarbital (Luminal) can be given for seizures lasting longer than 15 minutes (Victorio, 2022).
References
Adamolekun, B. (2022a). Drug treatment of seizures. Merck Manual Professional Version. https://www.merckmanuals.com/professional/neurologic-disorders/seizure-disorders/drug-treatment-of-seizures
Adamolekun, B. (2022b). Seizure disorders. Merck Manual Professional Version. https://www.merckmanuals.com/professional/neurologic-disorders/seizure-disorders/seizure-disorders
Centers for Disease Control and Prevention. (2020a). Epilepsy fast facts. https://www.cdc.gov/epilepsy/about/fast-facts.htm
Centers for Disease Control and Prevention. (2020b). Epilepsy: Types of seizures. https://www.cdc.gov/epilepsy/about/types-of-seizures.htm
Drislane, F. W. (2022). Convulsive status epilepticus in adults: Management. UpToDate. Retrieved March 9, 2023, from https://www.uptodate.com/contents/convulsive-status-epilepticus-in-adults-management
Epilepsy Foundation. (n.d.-a). Seizure triggers. Retrieved February 20, 2023, from https://www.epilepsy.com/what-is-epilepsy/seizure-triggers
Epilepsy Foundation. (n.d.-b). What is epilepsy? Retrieved February 20, 2023, from https://www.epilepsy.com/what-is-epilepsy
Epilepsy Foundation. (2013). Evaluating your medical history. https://www.epilepsy.com/diagnosis/medical-history
Epilepsy Foundation. (2017). Ketogenic diet. https://www.epilepsy.com/treatment/dietary-therapies/ketogenic-diet
Epilepsy Foundation. (2019a). Absence seizures. https://www.epilepsy.com/what-is-epilepsy/seizure-types/absence-seizures
Epilepsy Foundation. (2019b). Using seizure diaries. https://www.epilepsy.com/manage/tracking/seizure-diaries
Fick, D., Semla, T., Steinman, M., Beizer, J., Brandt, N., Dombrowski, R., DuBeau, C. E., Pezzulo, L., Eplin, J. J., Flanagan, N., Morden, E., Hanlon, J., Hollman, P., Linnebur., S., & Sandhu, S. (2012). American geriatrics society 2019 updated Beers criteria for potentially inappropriate medication use in older adults. Journal of American Geriatric Society, 67(4), 674-694. https://doi.org/10.1111/jgs.15767
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., Engel, J. Jr., Forsren, L., French, J. A., Glynn, M., Hesdorffer, D. C., Lee, B. I., Mathern, G. W., Moshe, S. L., Perucca, E., Scheffer, I. E., Tomson, T., Watanabe, M., & Wiebe, S. (2014). ILAE official report: A practical clinical definition of epilepsy. Epilepsia, 55(4), 475-482. https://doi.org/10.1111/epi.12550
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., Lagae, L., Moshe, S. L., Peltola, J., Perez, E. R., Scheffer, I. E., & Zuberi, S. M. (2017). Operational classification of seizure types by the international league against epilepsy: Position paper of the ILAE commission for classification and terminology. Epilepsia, 58(4), 522-530. https://doi.org/10.1111/epi.13670
Haider, H. A., & Bullinger, K. (2022). Neuroimaging in the evaluation of seizures and epilepsy. UpToDate. Retrieved February 20, 2023, from https://www.uptodate.com/contents/neuroimaging-in-the-evaluation-of-seizures-and-epilepsy
Huff, J. S., & Murr, N. (2022). Seizure. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK430765
Ignatavicius, D., Workman, L., & Rebar, C. (2018). Medical-surgical nursing: Concepts for interprofessional collaborative care (9th ed.). Elsevier.
International League Against Epilepsy. (2022a). Diagnostic manual: Dravet syndrome. https://www.epilepsydiagnosis.org/syndrome/dravet-overview.html
International League Against Epilepsy. (2022b). Diagnostic manual: Epilepsy by etiology. https://www.epilepsydiagnosis.org/aetiology/epilepsies-etiology-groupoverview.html
International League Against Epilepsy. (2022c). Diagnostic manual: Epilepsy classification. https://www.epilepsydiagnosis.org/epilepsy/epilepsy-classification-groupoverview.html
International League Against Epilepsy. (2022d). Diagnostic manual: Epilepsy syndromes. https://www.epilepsydiagnosis.org/syndrome/epilepsy-syndrome-groupoverview.html
International League Against Epilepsy. (2022e). Diagnostic manual: Focal onset seizure. https://www.epilepsydiagnosis.org/seizure/focal-seizure-overview.html
International League Against Epilepsy. (2022f). Diagnostic manual: Generalized onset seizure. https://www.epilepsydiagnosis.org/seizure/generalized-seizure-groupoverview.html
International League Against Epilepsy. (2022g). Diagnostic manual: Lennox Gastaut syndrome. https://www.epilepsydiagnosis.org/syndrome/lgs-overview.html
International League Against Epilepsy. (2022h). Diagnostic manual: Seizure classification. https://www.epilepsydiagnosis.org/seizure/seizure-classification-groupoverview.html
Karceski, S. (2022). Initial treatment of epilepsy in adults. UpToDate. Retrieved February 20, 2023, from https://www.uptodate.com/contents/initial-treatment-of-epilepsy-in-adults
Ko, D. Y. (2022). Epilepsy and seizures. https://emedicine.medscape.com/article/1184846-overview#a3
Mouhamed, Y., Vishnyakov, A., Qorri, B., Sambi, M., Frank, S. S., Nowierski, C., Lamba, A., Bhatti, U., & Szewczuk, M. (2018). Therapeutic potential of medicinal marijuana: An educational primer for health care professionals. Drug, Healthcare, and Patient Safety, 10, 45-66. https://doi.org/10.2147/dhps.s158592
OpenStax. (2018). Scans depicting CT, PET, and fMRI brain scans [Image]. https://commons.wikimedia.org/wiki/File:Scans_depicting_CT,_PET,_and_fMRI_brain_scans.jpg
Rosenberg, E. C., Tsien, R. W., Whalley, B. J., & Devinsky, O. (2015). Cannabinoids and epilepsy. Neurotherapeutics, 12(4), 747-768. https://doi.org/10.1007/s13311-015-0375-5
Schachter, S. C. (2022a). Evaluation and management of the first seizure in adults. UpToDate. Retrieved February 20, 2023, from https://www.uptodate.com/contents/evaluation-and-management-of-the-first-seizure-in-adults
Schachter, S. C. (2022b). Overview of the management of epilepsy in adults. UpToDate. Retrieved February 20, 2023, from https://www.uptodate.com/contents/overview-of-the-management-of-epilepsy-in-adults
Schachter, S. C. (2023). Antiseizure medications: Mechanisms of action, pharmacology, and adverse effects. UpToDate. Retrieved February 20, 2023, from https://www.uptodate.com/contents/antiseizure-medications-mechanism-of-action-pharmacology-and-adverse-effects
Tucker, R. (2022). 2022 Lippincott pocket drug guide for nurses (10th ed.). Wolters Kluwer.
US Food & Drug Administration. (2020). FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy. https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-comprised-active-ingredient-derived-marijuana-treat-rare-severe-forms
Victorio, M. C. (2022). Febrile seizures. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pediatrics/neurologic-disorders-in-children/febrile-seizures