About this course:
This course reviews the anatomy and physiology of the respiratory system and the epidemiology, risk factors, and causes of interstitial lung disease (ILD). It also covers the various classifications and subtypes of ILD, including the clinical manifestations, diagnosis, and treatment strategies for each type.
Course preview
Interstitial Lung Disease for APRNs
This course reviews the anatomy and physiology of the respiratory system and the epidemiology, risk factors, and causes of interstitial lung disease (ILD). It also covers the various classifications and subtypes of ILD, including the clinical manifestations, diagnosis, and treatment strategies for each type.
Upon completion of this module, learners should be able to:
- describe the anatomy and physiology of the respiratory system
- review the epidemiology, risk factors, and causes of ILD
- differentiate the four classifications of ILD, including the subtypes within each classification
- discuss the clinical manifestations, diagnosis, and treatment strategies for the various classification and subtypes of ILD
Interstitial lung disease, also known as diffuse parenchymal lung disease (DPLD), is an umbrella term used to describe a large heterogenous group of disorders that cause scarring of the lungs. This scarring, or fibrosis, causes lung stiffness, leading to difficulty breathing and impairs the lung's ability to carry oxygen. These lung diseases are grouped because of the similarities in their clinical, physiologic, pathological, and radiographic manifestations. The term interstitial can be confusing because it implies that the abnormalities are specific to the interstitium. However, many of these disorders involve a series of inflammation and fibrotic changes that extend beyond the disruption of the interstitial bed to changes in the parenchyma (i.e., alveoli, alveolar ducts, and bronchioles). The lung damage caused by ILDs is often progressive and irreversible (Antoine & Mlika, 2022; King, 2022a).
Anatomy and Pathophysiology of the Respiratory System
The respiratory system comprises the upper and lower respiratory tracts, which are responsible for moving air in and out of the lungs (see Figure 1). The upper respiratory tract warms and filters inspired air, while the lower respiratory tract is responsible for gas exchange. The upper respiratory tract includes the nose, paranasal sinuses, pharynx, tonsils, adenoids, larynx, and trachea. The nose provides a passageway for air to move to and from the lungs, filtering impurities and humidifying and warming the air during inhalation. As air enters the nostrils, the nasal mucosa (i.e., the large surface of moist, warm, highly vascular, ciliated mucous membranes) traps dust and organisms. The pharynx (throat) is a tubelike structure that connects the oral and nasal cavities to the larynx. The trachea serves as the passage between the larynx and the right and left main stem bronchi, which enter the lungs through an opening called the hilus (Hinkle & Cheever, 2018).
The lower respiratory tract contains the lungs, including the bronchial and alveolar structures needed for gas exchange. Gas exchange involves delivering oxygen through the bloodstream to the tissues and eliminating carbon dioxide during expiration. The lungs are paired, elastic structures enclosed in an airtight thoracic cage. The right lung is divided into the upper, middle, and lower lobes, while the left lung is divided into the upper and lower lobes. The lungs and the thoracic cavity are lined with a serous membrane (pleura) to provide lubrication, allowing smooth movement of the lungs within the thoracic cavity during inspiration and expiration. The bronchi within the lungs divide into the lobar, segmental, and subsegmental branches and the bronchioles. The lobar and segmental bronchi facilitate adequate postural drainage. The subsegmental bronchi are surrounded by connective tissue that contains arteries, lymphatics, and nerves. The bronchioles are the final branches in the lungs and contain no cartilage. Bronchiole patency depends on the elastic recoil of the surrounding smooth muscle and alveolar pressure. Bronchi and bronchioles contain cilia that propel mucus and foreign substances toward the larynx. Finally, the lungs comprise 300 million alveoli, including type I cells (providing a barrier between the air and the alveolar surface) and type II cells (producing surfactant). Surfactant reduces surface tension, allowing effective lung function (Hinkle & Cheever, 2018).
Figure 1
The Respiratory System
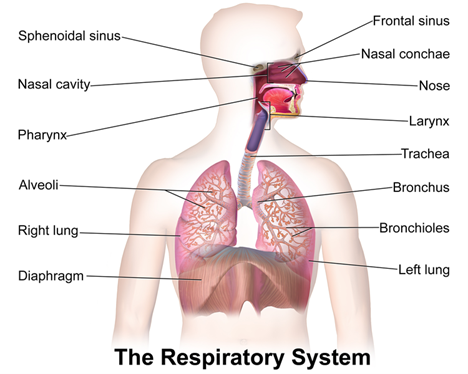
(BruceBlaus, 2013)
The respiratory system facilitates life-sustaining processes, including oxygen transport, respiration, ventilation, and gas exchange. Human cells rely on the oxidation of carbohydrates, fats, and proteins to produce energy. Without a continuous supply of oxygen, cells in the brain, heart, and other essential organs cannot survive. Oxygen is transported to, and carbon dioxide is removed from, the circulating blood through the thin walls of the capillaries. Oxygen diffuses through capillary walls to the interstitial fluid and eventually to the cells. Carbon dioxide diffuses in the opposite direction from the cells to the blood. After these tissue capillary exchanges, blood enters the systemic venous circulation and travels to the pulmonary circulation. The oxygen concentration in the alveoli is higher than in the blood; therefore, oxygen diffuses from the alveoli to the blood. Similarly, carbon dioxide has a higher concentration in the blood than in the alveoli, so it diffuses from the blood to the alveoli (see Figure 2; Hinkle & Cheever, 2018).
Figure 2
Human Gas Exchange
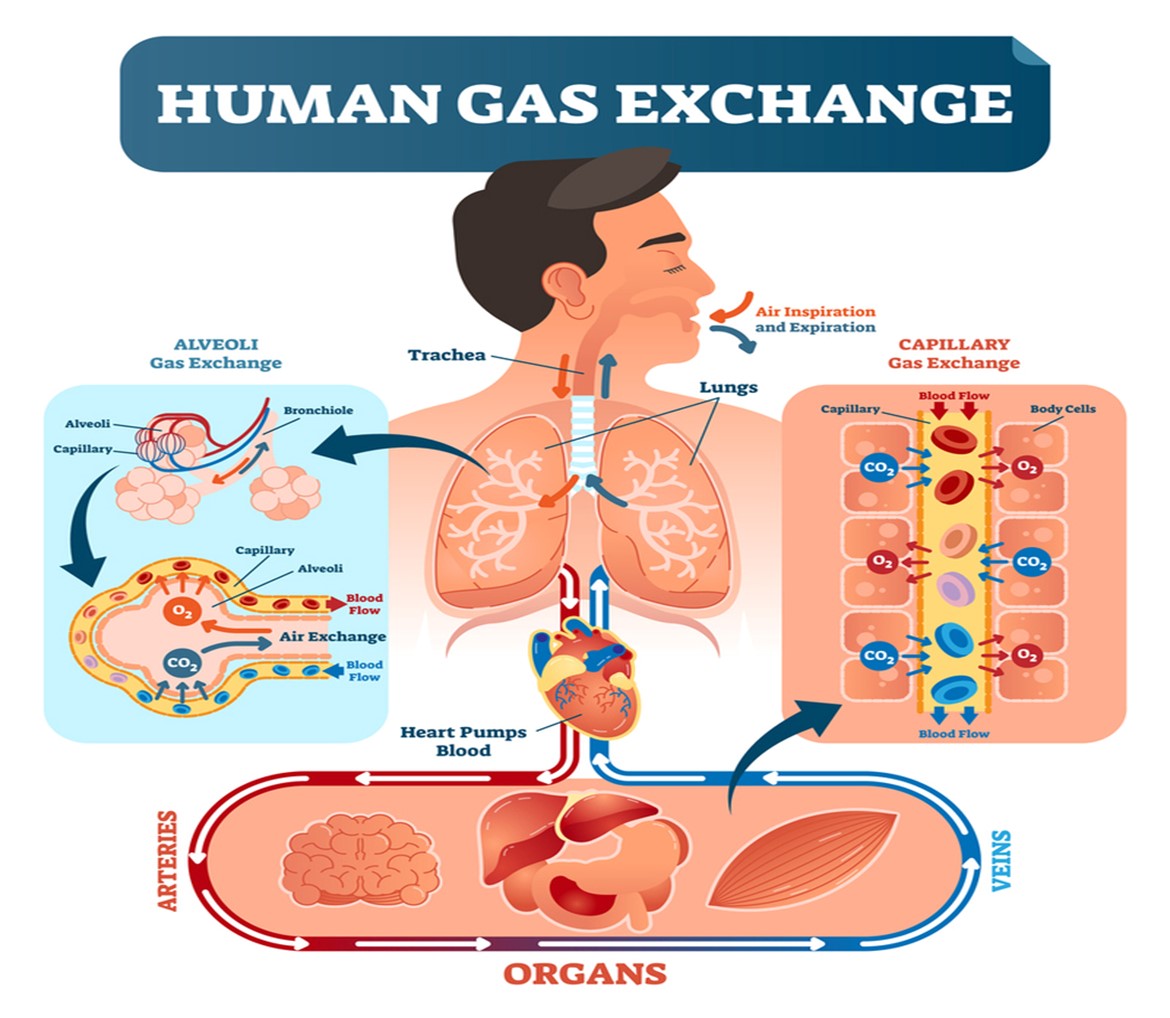
(Medic Tests, 2021)
Ventilation
Ventilation requires movement of the walls of the thoracic cage and the diaphragm. These movements alternate, increasing and decreasing the chest's capacity. When the chest capacity increases, air enters the trachea, passes through the bronchi and bronchioles, and inflates the alveoli in the lungs (inspiration). As the thoracic cavity expands, the pressure inside the thorax drops below the atmospheric pressure; air flows from a region of higher to lower pressure. During expiration, the diaphragm relaxes and the lungs recoil, decreasing the size of the thoracic cavity. As the pressure in the alveoli increases, air flows from the lungs to the atmosphere. The inspiratory phase of respiration requires active energy, while the expiratory phase is passive (Hinkle & Cheever, 2018).
Besides air-pressure variances, airway resistance, lung compliance, and lung volumes can also impact ventilation. The size or diameter of the airway, lung volumes, and airflow velocity determine airway resistance. Any process that changes the diameter of the airway will affect airway resistance and the rate of airflow. A more significant respiratory effort wil
...purchase below to continue the course
Perfusion
Gas exchange depends on pulmonary diffusion and perfusion. Pulmonary diffusion is how oxygen and carbon dioxide are exchanged in the body due to the differences in gas concentrations in the alveoli and capillaries. The alveolar-capillary membrane is ideal for diffusion due to its large, thin surface area, allowing for gas exchange from areas of high concentration to low concentration. Pulmonary perfusion refers to the blood flow through the pulmonary vasculature and is determined by pulmonary artery pressure, gravity, and alveolar pressure. The right ventricle pumps blood into the lungs through the pulmonary artery. The pulmonary artery supplies both lungs by dividing into the right and left branches. The normal range for systolic blood pressure in the pulmonary artery is about 20 to 30 mm Hg, and the diastolic pressure is 5 to 15 mm Hg, making pulmonary circulation a low-pressure system. Due to these lower pressures, the pulmonary vasculature can accommodate varying volumes of blood flow. Alveolar pressure also influences perfusion, as pulmonary capillaries lie between adjacent alveoli. Therefore, if the alveolar pressure is high, the capillaries are squeezed, impacting perfusion (Hinkle & Cheever, 2018).
Adequate gas exchange depends on effective ventilation and perfusion, resulting in an adequate ventilation-perfusion (V/Q) ratio. Four V/Q states can occur in the lungs: normal V/Q ratio, low V/Q ratio, high V/Q ratio, and absence of ventilation and perfusion (i.e., silent unit). In a healthy lung, the V/Q ratio is 1:1, indicating equal amounts of blood and gas moving through the alveoli. Low V/Q ratios (shunt) happen when perfusion (Q) exceeds ventilation (V). When blood bypasses the alveoli without gas exchange, shunting results in hypoxia, as the blood is delivered to the body's tissues without obtaining the necessary oxygen. Low V/Q ratios can occur with obstruction of the distal airways (e.g., tumor, atelectasis, pneumonia, mucus plug). High V/Q ratios (dead space) develop when ventilation exceeds perfusion, resulting in an inadequate blood supply for gas exchange in the alveoli (e.g., pulmonary emboli [PE], pulmonary infarction, cardiogenic shock). A silent unit occurs in the absence of ventilation and perfusion due to blockages (e.g., pneumothorax, ARDS; Hinkle & Cheever, 2018).
Restrictive Lung Disease
Lung compliance refers to the ease with which the lungs can be inflated or the change in lung volume can be achieved based on a given change in respiratory pressure. Noncompliant lungs are stiff and resistant to movement, requiring more pressure to move air than compliant lungs. The lung's collagen and elastin fibers, water content, and surface tension determine lung compliance. The compliance of the thoracic cage also plays a role in lung compliance. Lung tissue comprises elastin and collagen fibers, with elastin allowing the lungs to stretch easily with lung inflation. In contrast, collagen fibers resist stretching, making lung inflation more difficult. Lung compliance is decreased by conditions that reduce elastin, block bronchi or smaller airways, increase the surface tension of the alveoli, or impair the flexibility of the thoracic cage. With ILD, characteristic changes include alveolar septal thickening, fibroblast proliferation, and collagen deposition, which can lead to pulmonary fibrosis. As elastin fibers are replaced with scar tissue, lung compliance decreases (Martinez-Pitre et al., 2022; Norris, 2020).
Restrictive lung diseases are a heterogeneous group of disorders characterized by a reduced distensibility of the lungs (compromising lung expansion) and reduced lung volumes, specifically total lung capacity. Restrictive lung diseases, which are related to inflammatory and fibrotic changes, cause lung stiffening and decrease compliance. In contrast, obstructive lung diseases (e.g., asthma, emphysema) are caused by increased resistance to airflow due to partial or complete obstruction anywhere between the trachea and terminal bronchioles. Restrictive lung diseases that destroy the distal lung parenchyma (i.e., interstitium or interalveolar septa of the lung) due to inflammation, toxins, or other mechanisms are also ILDs (see Figure 3). Some ILDs can affect the distal part of the alveoli, causing restriction and decreased lung volumes. Other ILDs can affect the interstitium closer to the proximal aspect of the acinus near the bronchioles. This group of ILDs can cause obstruction but may not impact lung volumes. Although the causes of ILD can vary, they share a similar pattern of decreased lung capacity, diminished lung volumes, and varying degrees of hypoxia. ILDs may be acute or gradual in onset, progressing rapidly, progressing slowly, or remaining static (Lee, 2022n; Martinez-Pitre et al., 2022; Norris, 2020).
Figure 3
Interstitial Lung Disease
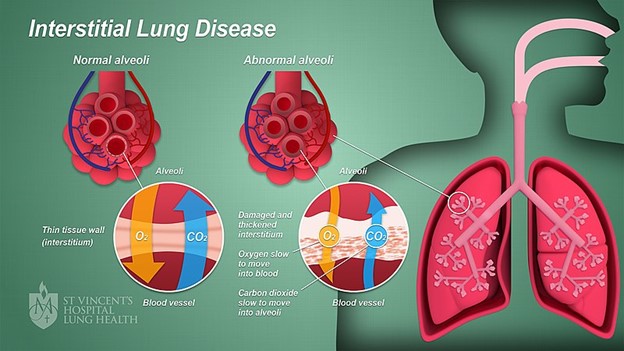
(Anatomyclassproj1, 2022)
Epidemiology
Pulmonary disorders are classified into obstructive pulmonary diseases and restrictive lung diseases. Obstructive pulmonary diseases are more common, accounting for 80% of all pulmonary disorders. In contrast, restrictive lung diseases are less common, only accounting for about 20% of all pulmonary disorders. The estimated incidence of ILD is 30 per 100,000 individuals, with a prevalence of 80.9 per 100,000 males and 67.2 per 100,000 females annually in the US. However, the true incidence and prevalence of ILD are challenging to determine and may be much higher. Since ILD is a diagnosis of exclusion, failure to recognize the disease has been challenging. As newer guidelines and classifications and more sophisticated diagnostic tools have become available, diagnosing ILD has become easier (Antione & Mlika, 2022; National Heart, Lung, and Blood Institute [NHLBI], 2022b).
Causes and Risk Factors
ILDs are characterized by damage to the lungs from environmental exposures, lifestyle habits, and other health conditions. These factors trigger an injury to the lungs; instead of the body repairing the tissue normally, an abnormal healing response occurs (i.e., alveoli become thick and scarred). Many different triggers can cause ILD, including airborne toxins, drugs, and medical treatments. However, in up to 30% of cases, the cause of ILD remains unknown (King, 2022a; Lee, 2022n; Norris, 2020). See Table 1 for a list of known causes of ILD.
Table 1
Causes of Interstitial Lung Disease
Category | Examples |
Occupational and environmental factors |
|
Drugs |
|
Connective tissue disorders |
|
Genetic disorders |
|
Vasculitis |
|
Idiopathic interstitial pneumonia (IIP) |
|
Miscellaneous disorders |
|
(King, 2022a; Lee, 2022n; Norris, 2020)
This list of possible causes of ILD provided in Table 1 is not exhaustive. Anyone can develop ILD, including children; however, certain factors increase the risk of developing ILD, with some being modifiable and others nonmodifiable. Risk factors for ILD include:
- age: ILD is more likely to affect adults than children
- exposure to occupational and environmental toxins: occupations such as mining, farming, and construction increase the risk of ILD
- gastroesophageal reflux disease (GERD): uncontrolled GERD increases the risk of ILD
- smoking: some forms of ILD are more likely to occur in individuals with a smoking history
- radiation and chemotherapy: a history of radiation and chemotherapy can increase the risk of ILD
- genetics: a family history of ILD increases the risk of developing ILD for close relatives
- gender: some ILDs are more common in men (e.g., IPF), while others are more common in women (e.g., LAM; King, 2022a; NHLBI, 2022a)
Clinical Evaluation
Patients with ILD will most commonly present with a cough or gradual onset of dyspnea. However, based on the heterogenous groups of disorders that comprise ILD, the clinical presentation can vary from pleuritic chest pain associated with sarcoidosis to hemoptysis associated with diffuse alveolar hemorrhages, or patients may be asymptomatic with abnormal chest imaging. The most specific presentations will be discussed below with the various classifications of ILD. Given the different subtypes of ILD, a thorough review and documentation of each individual's past medical history are important to identify the causative factor of the suspected ILD (Antoine & Mlika, 2022; King, 2022a; Norris, 2020).
A thorough history should include the following (Antoine & Mlika, 2022; King, 2022a; Norris, 2020):
- Onset of symptoms: The duration of symptoms can help differentiate between acute and subacute ILD presentation (e.g., acute eosinophilic pneumonia, connective tissue associated-ILD) and chronic presentation (e.g., sarcoidosis, pneumoconioses, IPF).
- Age and gender: Some ILDs are more prevalent in men, while others are more prevalent in women; individuals over 50 are more likely to have IPF, while other ILDs are more likely to affect individuals aged 20 to 40 (e.g., sarcoidosis, connective tissue associated-ILD, inherited ILD, pulmonary Langerhans cell histiocytosis, and lymphangioleiomyomatosis).
- Past medical history: A history of certain diseases (e.g., connective tissue disease, malignancy, inflammatory bowel disease [IBD]) or medications used to treat those diseases can increase the risk of ILD. Asthma and allergic rhinitis are linked to chronic eosinophilic pneumonia, and an immunocompromised state may suggest an infectious cause of ILD.
- Smoking: A history of smoking is important, as some ILDs are more common in nonsmokers or former smokers (e.g., sarcoidosis and hypersensitivity pneumonitis), and others are more prevalent in smokers (e.g., IPF, pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonitis, respiratory-bronchiolitis-ILD).
- Family history: A family history may help identify some ILDs with a genetic component (e.g., IPF, nonspecific interstitial pneumonia, sarcoidosis).
- Medication use and irradiation: A review of all medications (i.e., prescription, over-the-counter, supplements) taken and any exposure to irradiation (i.e., dose and duration) can help exclude drug-induced or radiation-induced ILD. ILDs related to drugs or radiation can occur weeks, months, or even years after exposure.
- Occupational and environmental exposures: A review of the home and work environments can help identify exposures that increase the risk of ILD. Healthcare providers (HCPs) should also ask about the duration and degree of exposure and whether protective devices were used.
- Symptoms: HCPs should assess symptoms, including their duration, progression, and severity. Common symptoms of ILDs include dyspnea and coughing (usually dry). Hemoptysis may occur in lymphangioleiomyomatosis, diffuse alveolar hemorrhage syndromes, and tuberous sclerosis. Wheezing is uncommon in ILDs but may be seen with lymphangitic carcinomatosis, respiratory bronchiolitis, and chronic eosinophilic pneumonia. Chest pain is also uncommon but may be present with SLE, mixed connective tissue disease, rheumatoid arthritis, and some drug-induced ILDs. Extrapulmonary symptoms may also accompany connective tissue diseases, including fever, joint pain, weakness, pleuritis, dry eyes, and dry mouth.
Physical Examination
The physical examination of patients with ILD is usually nonspecific, except in the case of systemic diseases when there are extrapulmonary findings. Most patients with ILD will have crackles during lung auscultation, even without radiographic abnormalities. Although crackles are less likely to be heard in patients with granulomatous disease. The cardiac examination is usually unremarkable in patients with ILD. Some patients with advanced disease could have signs of pulmonary hypertension, including peripheral edema, accentuated p2 heart sounds, or a right ventricular heave. Clubbing of the nails is common with some ILDs (e.g., IPF) and rare in others (e.g., hypersensitivity pneumonitis, sarcoidosis, pulmonary Langerhans cell histiocytosis). When clubbing is present, it is usually a late sign indicating fibrosis of the lungs (Antoine & Mlika, 2022; King, 2022a; Norris, 2020).
Laboratory Testing
The general laboratory testing for suspected ILDs usually includes a complete blood count (CBC) with differential to evaluate for anemia, leukocytosis, eosinophilia, and polycythemia. A basic metabolic profile (BMP) to assess kidney function and liver function tests (LTFs) should be ordered. Serological studies should also be ordered to identify rheumatic diseases (e.g., anti-nuclear antibodies [ANA], rheumatoid factor). Additional serological testing may be warranted for patients with a positive ANA test. Serological testing for hypersensitivity pneumonitis antibodies should be done based on known patient exposure (Antoine & Mlika, 2022; King, 2022a).
Imaging
Imaging evaluation should start with a chest radiograph. The reticular pattern (i.e., a collection of small linear opacities forming a net-like appearance) is most commonly seen on chest radiography. However, nodular or mixed patterns (i.e., alveolar filling and increased interstitial markings) may be present. Whenever possible, a chest radiograph should be compared to previous images to determine the rate of change in disease activity. HCPs should be aware that 10% of chest radiographs will appear normal in some forms of ILD, especially hypersensitivity pneumonitis. Although a chest radiograph helps identify suspected ILD, high-resolution computed tomography (HRCT) offers a more accurate diagnosis and characterization of ILD. HRCT images should be tailored specifically to ILD evaluation, with imaging of the entire chest with the patient supine at sustained end-inspiration and then sustained end-expiration (Antoine & Mlika, 2022; King, 2022a).
Pulmonary Function Testing
Pulmonary function testing (i.e., lung volumes, spirometry, diffusing capacity) should be performed on patients with suspected ILD. Lung function can help HCPs assess the severity of respiratory impairment and determine if the changes are obstructive, restrictive, or a mixed pattern. Most ILDs have a restrictive pattern that reduces the total lung capacity (TLC), functional residual capacity (FRC), and residual volume (RV). The forced expiratory volume in 1 minute (FEV1) and the forced vital capacity (FVC) decrease. However, these changes are proportional to decreased lung volumes, resulting in a normal or increased FEV1/FVC ratio. Reduced lung volumes worsen as lung stiffness increases with disease progression. Some ILDs have an obstructive pattern (e.g., sarcoidosis, hypersensitivity pneumonitis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis), which reduces FEV1/FVC ratio (Antoine & Mlika, 2022; King, 2022a).
Diffusing capacity is considered a nonspecific finding with ILD. A decrease in diffusing capacity is common due to the effacement of the alveolar-capillary units and the extent of mismatching of the ventilation and perfusion of the alveoli. A moderate to severe reduction in diffusing capacity with normal lung volumes could indicate pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, or combined ILD with emphysema or pulmonary vascular disease. A resting and exercise pulse oximetry should be measured, and arterial blood gases (ABG) are often obtained to corroborate the results. A normal resting ABG may be present in early ILD or may reveal hypoxemia and respiratory alkalosis. HCPs should know that normal resting pulse oximetry and ABG do not rule out significant hypoxemia during sleep or exercise. Exercise testing can be done using a cardiopulmonary exercise test (i.e., performed on a cycle ergometer or treadmill to measure heart rate, pulse oximetry, and electrocardiography) or a 6-minute walk test. Serial assessment of resting and exercise gas exchange helps evaluate the severity of ILD and its responsiveness to treatment. Pulse oximetry desaturation below 88% in a 6-minute walk test is associated with a median survival of 3.21 years compared to 6.63 years for patients who do not experience desaturation below 88% (Antoine & Mlika, 2022; King, 2022a).
Cardiac Evaluation
An electrocardiogram (ECG) should be obtained during the initial evaluation of ILD to rule out concurrent cardiovascular disease or pulmonary hypertension. If pulmonary hypertension or heart failure is suspected, levels of serum brain natriuretic peptide (BNP) or N-terminal-proBNP should be obtained. Further cardiac testing, including an echocardiogram, should be done for an abnormal ECG, rapid-onset symptoms, suspected heart failure, signs of pulmonary hypertension (e.g., a moderate to severe reduction in diffusing capacity or abnormal heart rate recovery after a 6-minute walk test), or before performing a lung biopsy (Antoine & Mlika, 2022; King, 2022a).
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) can be used to evaluate ILD associated with hemoptysis (to determine an alveolar source of bleeding or infectious etiology) or acute or rapid onset (to determine malignancy or acute eosinophilic pneumonia). BAL can also be used for a subacute or chronic presentation to help differentiate the type of ILD when hypersensitivity pneumonitis, pulmonary Langerhans cell histiocytosis, infection, or sarcoidosis are suspected. However, tissue samples are often needed for a formal diagnosis. BAL is performed during a flexible bronchoscopy to obtain fluid and cells from the distal airways and alveoli to be evaluated for cell counts, cytologic analysis, or mycobacterial, fungal, or viral cultures. For patients with suspected IPF, BAL is less likely to be helpful (Antoine & Mlika, 2022; King, 2022a).
Lung Biopsy
A lung biopsy should be performed when the results of the previously discussed evaluation methods do not provide a confident diagnosis. HCPs should consider lung biopsy on a case-by-case basis in terms of the likelihood of diagnosis and the risk of complications. Indications for lung biopsy can include atypical or sudden changes in radiographic features, rapid clinical deterioration, unexplained extrapulmonary manifestations, or atypical or progressive symptoms (e.g., hemoptysis, fever, weight loss). In addition, a lung biopsy may be indicated to exclude neoplastic or infectious processes. HCPs should collaboratively discuss options with patients based on symptoms, physiologic impairment, and radiographic abnormalities. For example, some patients may prefer to monitor symptoms with interval pulmonary function tests, while others prefer a definitive diagnosis. A lung biopsy can be performed by video-assisted thoracoscopic biopsy, flexible bronchoscopy, or thoracotomy (Antoine & Mlika, 2022; King, 2022a).
Classification and Management Strategies
ILDs can be classified in various ways, including acute versus chronic, known cause versus unknown cause, granulomatous versus non-granulomatous, primary lung disease versus secondary to systemic disease, and associated with a history of smoking versus no smoking history. ILD is usually broken into four primary classifications, with some subcategories within IIP (King, 2022a; Lee, 2022n):
- DPLD with a known cause (e.g., drugs, environmental, or triggers such as rheumatic disease)
- IIP, with 20% being familial and 80% nonfamilial
- chronic fibrosing (e.g., IPF, idiopathic nonspecific interstitial pneumonia)
- acute/subacute fibrosing (e.g., cryptogenic organizing pneumonia, acute interstitial pneumonia)
- smoking-related (e.g., respiratory bronchiolitis interstitial lung disease, desquamative interstitial pneumonia)
- Granulomatous DPLD (e.g., sarcoidosis)
- Other forms of DPLD (e.g., LAM, pulmonary Langerhans cell histiocytosis [PLCH], eosinophilic pneumonia)
DPLD with a Known Cause: Occupational and Environmental Interstitial Lung Disease
Occupational and environmental ILDs include drug-induced ILD, pneumoconioses, and hypersensitivity diseases. Pneumoconioses are caused by inhaling inorganic dust, while hypersensitivity diseases are caused by inhaling organic dust related to occupational antigens. Byssinosis is a third type of occupational lung disease that affects cotton workers and has characteristics of pneumoconioses and hypersensitivity lung diseases. The development of pneumoconioses depends on the particle size, chemical nature, concentration, and length of exposure. When particles enter the lungs, macrophages engulf and transport the particles away from the small bronchioles and alveoli to be cleared from the lungs. The effectiveness of macrophage activity can be impacted by smoking, consumption of alcohol, and hypersensitivity reactions. The ingestion of silica particles (i.e., silicosis) causes the destruction of macrophages and the release of substances that cause inflammation and fibrosis. Due to the destruction of macrophages, individuals with silicosis are at increased risk of tuberculosis. The treatment for occupational and environmental ILDs includes stopping the exposure (Antoine & Mlika, 2022; Norris, 2020).
Drug-induced pulmonary diseases occur when an individual without previous pulmonary disease develops respiratory symptoms, deterioration of pulmonary function, and radiographic or histologic changes. An estimated 150 drugs or more have caused pulmonary disease, possibly due to hypersensitivity reactions, although the exact mechanism of action varies and is often not well understood. Drug-induced reactions can be acute or chronic and typically affect the parenchyma, resulting in interstitial changes on x-ray. The histopathology of drug-induced reactions can vary, but hypersensitivity pneumonitis and eosinophilic pneumonia are often seen. Treatment for drug-induced ILD includes stopping the drug that is causing pulmonary disease (Lee, 2022f; Norris, 2020).
Hypersensitivity Pneumonitis
Hypersensitivity occupational lung diseases (e.g., hypersensitivity pneumonitis) are caused by prolonged, intense exposure to inhaled organic dust (e.g., farmer's lung due to moldy hay; pigeon breeder's lung due to excreta, serum, or feathers of birds; humidifier or air conditioner lung due to mold in water reservoirs; and bagassosis due to contaminated sugar cane). A hypersensitivity reaction primarily involves the alveoli and can cause progressive pulmonary fibrosis. Repeated exposure to the antigen leads to acute neutrophilic and mononuclear alveolitis, prompting interstitial lymphocytic infiltration and granulomatous reaction. Cigarette smoking may delay or prevent the development of hypersensitivity pneumonitis but exacerbates the disease once it occurs. Hypersensitivity pneumonitis can be acute (i.e., dyspnea 4 to 8 hours after exposure), occurring in previously sensitized individuals who experience an acute high-level antigen exposure. Signs and symptoms include coughing, fever, chest tightness, and inspiratory crackles. Chronic hypersensitivity pneumonitis affects individuals with long-term low-level antigen exposure, with symptoms manifesting over months to years. Chronic disease symptoms include fatigue, exertional dyspnea, a productive cough, and weight loss; individuals with advanced cases can experience right sided-heart failure and pulmonary fibrosis. Subacute hypersensitivity pneumonitis develops over days to weeks, with acute symptoms superimposed on chronic symptoms (King, 2022b; Lee, 2022g).
A diagnosis of hypersensitivity pneumonitis is based on a high index of suspicion in patients with symptoms and an identified exposure. It should always be considered for patients with newly identified ILD. A chest radiograph is often normal in acute and subacute forms, but reticular or nodular opacities can be present. Chronic disease is more likely to have chest radiograph abnormalities, including reticular or nodular opacities in the upper lobes, reduced lung volumes, and honeycombing (i.e., small cystic spaces with irregularly thickened walls), similar to IPF. HRCT is the gold standard for evaluating parenchymal changes and diagnosing hypersensitivity pneumonitis. Pulmonary function tests should also be completed. Hypersensitivity pneumonitis can cause obstructive, restrictive, or mixed airway changes, with chronic disease most commonly causing restrictive changes. Bronchoalveolar lavage is often nonspecific for hypersensitivity pneumonitis, and surgical lung biopsy should be considered if noninvasive testing is inconclusive (King, 2022b; Lee, 2022g).
Treatment should include eliminating antigen exposure; acute disease is often reversible if detected early and after the exposure is eliminated. Treatment of acute and subacute hypersensitivity pneumonitis includes corticosteroids, usually prednisone (Deltasone). Dosing should be 60 mg orally once a day for 1 to 2 weeks, followed by a taper to 20 mg daily over 2 to 4 weeks, followed by a decrease of 2.5 mg weekly until the drug is stopped. Treatment of chronic disease requires a longer course of prednisone. Dosing in these cases is 30 mg to 40 mg orally daily, with tapering dependent on the clinical course. Side effects of prednisone (Deltasone) include insomnia, increased appetite, weight gain, acne, headaches, dizziness, nausea, and slow wound healing. Long-term use of prednisone (Deltasone) weakens the immune system, and patients should be educated on avoiding close contact with sick individuals. HCPs should also teach patients about the importance of tapering prednisone (Deltasone) and not stopping abruptly because of the risk of withdrawal symptoms (e.g., fatigue, weakness, body aches, nausea, vomiting). Some patients may require corticosteroid-sparing agents (e.g., mycophenolate [Rheumatrex], azathioprine [Imuran]) for long-term treatment (Lee, 2022g).
Idiopathic Interstitial Pneumonia
IIP is a subtype of ILD of unknown etiology characterized by similar clinical and radiographic features. There are eight histologic subtypes of IIP that are characterized by varying degrees of interstitial inflammation and fibrosis, as distinguished by the histopathologic patterns found on lung biopsy. Symptoms of IIPs are usually nonspecific and include dyspnea on exertion and a cough that varies in onset and progression. HCPs may observe reduced chest expansion, bibasilar end-expiratory crackles, tachypnea, and clubbing. IIPs should be suspected of any patient with unexplained ILD. A chest radiograph is typically abnormal but not specific to differentiate between subtypes. Pulmonary function tests are also helpful in differentiating between subtypes but cannot classify the severity of impairment. IIPs are typically restrictive, with reduced lung volumes and diffusion capacity. Hypoxemia is also common during rest and exercise (Cool, 2021; Lee, 2022m).
HRCT should be done for all patients with suspected IIP. The patient should be supine and prone and should include dynamic expiratory imaging to assess for small airway involvement. HRCT can provide information on the potential etiology and distribution of disease and identify an underlying or coexisting disease. Serologic tests should be obtained for patients whose clinical features suggest a connective tissue disorder. Although bronchoscopic transbronchial biopsy helps differentiate some ILDs, it does not yield enough tissue to diagnose IIPs. Bronchoalveolar lavage can provide information about a patient's disease progression and response to therapy and help narrow the differential diagnosis for some patients. A lung biopsy is needed when the history, clinical features, and HRCT cannot confirm a diagnosis. A biopsy of multiple sites with video-assisted thorascopic surgery (VATS) is recommended. Treatment for IIPs will vary by subtype but can include corticosteroids and sometimes lung transplantation (Cool, 2021; Lee, 2022m).
Idiopathic Pulmonary Fibrosis
IPF is the most common subtype of IIP, with men being affected twice as often as women. IPF also has a genetic predisposition, with familial clustering in approximately 20% of cases. There is a strong association between IPF in current and former smokers. Histologically, IPF is identified as interstitial pneumonia, with sites of fibroblast proliferation and dense scarring alternating with normal lung tissue. In addition, scattered interstitial inflammation occurs with plasma cell, lymphocyte, and histiocyte inflammation. Honeycombing is present in all patients and worsens with advanced disease. Symptoms of IPF include a nonproductive cough, clubbing, and dyspnea on exertion that develop over 6 months to several years. Fine, dry, inspiratory crackles (i.e., Velcro crackles) at both bases are a hallmark of IPF. Unfortunately, signs of IPF can be missed because of the similarities to asthma, bronchitis, and heart failure (Cool, 2021; Lee, 2022i).
Chest radiography typically shows diffuse reticular opacities in the lower and peripheral lung fields, dilated arteries, and honeycombing. An IPF diagnosis requires HRCT showing a usual interstitial pneumonia pattern characterized by diffuse, patchy, subpleural, and reticular opacities. Irregularly thickened intralobular lines, interlobular septa, and honeycombing will be present. Ground-glass opacities (i.e., areas of hazy increased attenuation of the lungs) suggest an alternative diagnosis. The median life expectancy of IPF is about 3 years. Antifibrotic drugs (e.g., pirfenidone [Esbriet], nintedanib [Ofev]) can slow the progression of IPF (i.e., declining lung function) and reduce the risk of acute respiratory deterioration, which increase morbidity and mortality. Additional supportive measures include oxygen and pulmonary rehabilitation. Lung transplantation is also an option for otherwise healthy individuals, generally for those younger than 65 (Cool, 2021; Lee, 2022i).
Desquamative Interstitial Pneumonia
Desquamative interstitial pneumonia is a subtype of IIP almost exclusively found in current and former smokers, who tend to develop the disease between ages 30 and 40. Desquamative interstitial pneumonia is characterized by cuboidal pneumocytes that line the alveolar walls, with moderate infiltration of the alveolar septum by plasma cells and lymphocytes. These lung changes typically occur uniformly in the lung parenchyma. Desquamative interstitial pneumonia is a chronic lung infection, manifesting gradually with a nonproductive cough and increasing dyspnea. Chest radiography typically shows bibasilar ground-glass opacities; however, up to 20% of cases have a normal radiograph. HRCT is necessary to diagnose desquamative interstitial pneumonia. This imaging typically shows multifocal or diffuse, basilar, subpleural ground-glass opacities; cysts can be present where ground-glass opacities are found. Irregular linear and reticular opacities may also be seen but are not the most common features of desquamative interstitial pneumonia. A lung biopsy may be required if HRCT is inconclusive. Smoking cessation is the hallmark management strategy, with a 75% improvement in patients who quit. If smoking cessation does not resolve symptoms, systemic corticosteroid therapy (prednisone [Deltasone]) over months has been the most effective pharmacologic intervention. In addition to systemic corticosteroids, patients can also be given immunosuppressive therapy, such as azathioprine (Imuran). This combination is effective for most patients, with some stabilizing and others with complete recovery. Desquamative interstitial pneumonia has a 70% survival at 10 years (Cool, 2021; Lee, 2022e).
Nonspecific Interstitial Pneumonia
Nonspecific interstitial pneumonia is a subtype of IPP that mainly affects nonsmokers, women, and individuals over 50. Although the clinical presentation of nonspecific interstitial pneumonia is similar to IPF, it is much less common. Most cases of nonspecific interstitial pneumonia have no known cause or association. However, they have a pathologic process that resembles connective tissue disorders, hypersensitivity pneumonitis, and drug-induced pulmonary disease. Patients usually present with coughing and dyspnea that has been present for months to years. Nonspecific interstitial pneumonia is a diagnosis of exclusion and requires HRCT and lung biopsy to rule out alternative disorders (e.g., connective tissue disorders, drug toxicities, or hypersensitivity pneumonitis). Chest radiography typically shows reticular opacities in the lower lobes. HRCT findings often include bilateral, patchy ground-glass attenuation, irregular lines, and bronchial dilation in the lower lung regions. A surgical lung biopsy is required for a nonspecific interstitial pneumonia diagnosis. Histologically, there will be temporally homogeneous inflammation and fibrosis, in contrast to heterogeneity in usual interstitial pneumonia. The prognosis depends on the degree of fibrosis—nearly 100% survival at 10 years for mild cases and a median survival of 3 to 5 years for severe cases. Patients typically respond to treatment with corticosteroids, with or without immunosuppressive drugs such as mycophenolate (Rheumatrex), azathioprine (Imuran), cyclophosphamide (Cytoxan; Cool, 2021; Lee, 2022l).
Cryptogenic Organizing Pneumonia
Cryptogenic organizing pneumonia is a subtype of IIP characterized by granulation tissue obstructing alveolar ducts and spaces, with chronic inflammation occurring in adjacent alveoli. Cryptogenic organizing pneumonia does not appear to be related to smoking and affects men and women equally, usually between ages 40 and 50. The clinical presentation typically includes a progressive cough and exertional dyspnea with velcro-like crackles. Approximately 50% of patients report having a recent flu-like illness characterized by coughing, fever, fatigue, malaise, and weight loss. Chest radiography typically shows diffuse, bilateral, peripherally distributed alveolar opacities with normal lung volumes. Unilateral opacities, irregular linear or nodular opacities, and honeycombing are rarely seen. HRCT findings include patchy airspace consolidation in the peripheral and lower lung regions (in 90% of cases), small nodular opacities, ground-glass opacities, and bronchial wall thickening and dilation. Pulmonary function tests usually show restrictive abnormalities, but these tests can also be normal or show obstructive abnormalities (21% of cases). Bronchoscopic or surgical lung biopsy results show excessive proliferation of granulation tissue within alveolar ducts and small airways. There will also be chronic inflammation in the surrounding alveoli. Corticosteroids are the treatment of choice for cryptogenic organizing pneumonia. Disease recurrence affects up to 50% of patients; therefore, treatment is usually given for 6 to 12 months, even though symptoms typically resolve in 2 weeks (Cool, 2021; Lee, 2022d).
Respiratory Bronchiolitis-Associated Interstitial Lung Disease
Respiratory bronchiolitis-associated interstitial lung disease (RBILD) is a subtype of IIP characterized by submucosal inflammation of the membranous and respiratory bronchioles, mucus stasis, tan-brown pigmented macrophages, cuboidal epithelium in the bronchioles and alveoli, and alveolar septal scarring. RBILD occurs primarily in smokers and is twice as common in males than in females. Signs and symptoms include dyspnea on exertion, coughing, and crackles in the lungs. Chest radiographic findings consist of small regular and irregular opacities; diffuse, fine reticular or nodular opacities; bronchial wall thickening; small peripheral rings; and prominent peribronchovascular interstitium. HRCT results show centrilobular nodules and patchy areas of hazy ground-glass opacities. Routine laboratory testing does not help make a diagnosis. Pulmonary function tests can show a mixed restrictive-obstructive pattern. A surgical lung biopsy is recommended if a smoking cessation trial does not decrease symptoms and the diagnosis remains unclear. Smoking cessation and avoidance of passive cigarette smoke are the recommended treatment. There is minimal evidence to support the use of corticosteroids for RBILD (Cool, 2021; Lee, 2022p).
Acute Interstitial Pneumonia
Acute interstitial pneumonia (AIP) is a rare subtype of IIP that manifests similarly to acute respiratory distress syndrome (ARDS), usually occurring in individuals over 40. It is equally prevalent among males and females. AIP is characterized by organizing diffuse alveolar damage, marked alveolar septal edema with inflammatory cell proliferation, thickening of the alveolar wall, and fibroblast proliferation. In addition, septa are lined with atypical, hyperplastic type II pneumocytes, airspaces are collapsed, and thrombi develop in small arteries. Clinical presentation includes coughing, shortness of breath, and a fever that increases in severity over 7 to 14 days, progressing to respiratory failure. Chest radiography findings include diffuse bilateral airspace opacification. HRCT findings typically show bilateral patchy symmetric areas of ground-glass attenuation and bilateral areas of airspace consolidation, mainly with a subpleural distribution. A surgical lung biopsy is usually required for a definitive diagnosis due to the similarities with acute IPF, diffuse alveolar hemorrhage, acute eosinophilic pneumonia, and cryptogenic organizing pneumonia. These findings include diffuse alveolar damage without a known cause of ARDS. Patients with AIP are often critically ill and require support treatment measures, including mechanical ventilation (MV). Corticosteroids are often used, but their efficacy has not been well-established for AIP. AIP has a poor prognosis, with more than 60% of patients dying within 6 months due to respiratory failure (Cool, 2021; Lee, 2022b).
Lymphocytic Interstitial Pneumonia
Lymphocytic interstitial pneumonia (LIP) is a rare subtype of IIP characterized by infiltration of alveoli and alveolar septa with small lymphocytes and varying numbers of plasma cells. It is hypothesized that LIP is caused by an autoimmune disease (i.e., Sjogren syndrome, SLE, rheumatoid arthritis, Hashimoto thyroiditis) or a reaction to a viral infection (e.g., Epstein-Barr virus). LIP is the most common cause of pulmonary disease after a pneumocystis infection in children with human immunodeficiency virus (HIV). Clinical manifestations of LIP include progressive (months to years) dyspnea and coughing, with a mean age of appearance of 54. Other symptoms include night sweats, arthralgias, fever, and weight loss. Crackles may be heard on lung examination. LIP diagnosis is suspected in at-risk patients with consistent symptoms (Cool, 2021; Lee, 2022k).
Chest radiography shows bibasilar linear reticular or nodular opacities, with alveolar opacities and cysts in more advanced disease. HRCT should be done to define the hilar anatomy, identify pleural involvement, and determine the extent of the disease. However, the findings can be highly variable and include centrilobular and subpleural nodules, nodular ground-glass opacities, cystic structures, and thickened bronchovascular bundles. Bronchoalveolar lavage is done to rule out infection, and serum protein electrophoresis is needed because 80% of cases have a serum protein abnormality. A lung biopsy is required for diagnosis, demonstrating expansion of the alveolar septa due to lymphocytic and other immune cell infiltrates. Immunohistochemical staining and flow cytometry must be completed to distinguish between LIP (polyclonal infiltrates) and primary lymphoma (monoclonal infiltrates). Treatment includes corticosteroids and cytotoxic drugs. The 5-year survival rate is 50% to 66%, with death occurring due to pulmonary fibrosis or the development of malignant lymphoma (Cool, 2021; Lee, 2022k).
Idiopathic Pleuroparenchymal Fibroelastosis
Idiopathic pleuroparenchymal fibroelastosis is a rare subtype of IIP that involves the slow progression of upper lobe fibrosis of the pleura and subpleural lung parenchymal. Most patients are nonsmokers, and the median age at presentation is 57. Patients often report a history of recurrent infections, dry coughing, and shortness of breath; pneumothorax is also common with idiopathic pleuroparenchymal fibroelastosis. Chest radiography and HRCT findings include upper lobe thickening of the pleura and subpleural regions. Some patients may experience co-existent interstitial pneumonia in the lower lobes. A surgical lung biopsy is required for diagnosis. Treatment can include corticosteroids, but disease progression occurs in 60% of cases (Cool, 2021; Lee, 2022h).
Granulomatous DPLD: Sarcoidosis
Granulomatous DPLD is the third category of ILD, with sarcoidosis being the most common of all ILDs. Sarcoidosis is a multisystem inflammatory disease that frequently begins between ages 20 and 40 and is more prevalent in Black Americans and those with northern European heritage. Sarcoidosis is a cell-mediated immune response to an environmental antigen in a genetically susceptible person characterized by the accumulation of T cells and macrophages, the release of chemokines and cytokines, and the organization of responding cells into granulomas. Granulomas are collections of mononuclear cells and macrophages that differentiate into epithelioid and multinucleated giant cells and are surrounded by plasma cells, collagen, lymphocytes, and fibroblasts. Granulomas most commonly occur in the lymph nodes and the lungs, causing significant destruction and fibrosis. Within the lungs, granulomas are distributed along lymphatics, most commonly in the subpleural, peribronchiolar, and perilobular regions. Signs and symptoms of sarcoidosis include chest discomfort, coughing, dyspnea, and crackles. Other symptoms consist of fever, weakness, weight loss, and malaise (Iannuzzi, 2022).
Bilateral hilar adenopathy is the most common abnormality found on a chest radiograph; however, chest radiographs are poor predictors of pulmonary function and disease severity. HRCT is more sensitive for detecting hilar and mediastinal lymphadenopathy and parenchymal abnormalities. Advanced sarcoidosis findings include beading of the interlobular septa; traction bronchiectasis; ground-glass opacities; parenchymal nodules, cysts, or cavities; and thickening of the bronchovascular bundles and bronchial walls. A definitive diagnosis is confirmed by noncaseating granulomas obtained from a lung biopsy. Endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) is the diagnostic procedure of choice, detecting sarcoidosis in 90% of biopsies of the mediastinal or hilar lymph nodes. Pulmonary function tests are often normal in early sarcoidosis, showing restriction and reduced diffusing capacity in advanced disease. Patients with extensive lung involvement may have a normal oxygen saturation at rest but desaturate with exertion (Iannuzzi, 2022).
The disease severity of sarcoidosis is highly variable, with approximately 66% of cases achieving remission with few or no sequelae. Sarcoidosis is considered chronic if remission does not occur within 2 to 3 years, a feature occurring in 30% of patients. Approximately 10% to 20% of people experience permanent sequelae, and the disease is fatal in 1% to 5% of cases due to respiratory failure caused by pulmonary fibrosis. Since sarcoidosis often resolves spontaneously, patients who are asymptomatic or have mild symptoms do not require treatment. These patients should be followed with serial chest radiographs and pulmonary function tests (Iannuzzi, 2022).
Treatment with corticosteroids is required for patients with worsening symptoms, deteriorating lung function, or worrisome changes on radiography. Prednisone (Deltasone) 20 mg to 40 mg daily is the standard protocol for sarcoidosis treatment. A response to treatment usually takes 6 to 12 weeks, and prednisone (Deltasone) can be tapered to 10 mg to 15 mg daily for 6 to 9 months with symptom improvement. Immunosuppressants can be used for patients who cannot tolerate prednisone (Deltasone) or in cases that are refractory to moderate to high doses of prednisone (Deltasone). Methotrexate (Rheumatrex) is most commonly used, with a 6-month trial of 10 mg to 15 mg orally per week. Initially, methotrexate (Rheumatrex) and corticosteroids are given together for 6 to 8 weeks, and then the corticosteroid can be tapered and eventually stopped. Patients can take 6 to 12 months to achieve the maximal response with methotrexate (Rheumatrex), and the corticosteroid may need to be tapered more slowly. Patients should be tested for hepatitis B and C before starting methotrexate (Rheumatrex), and serial blood counts and liver enzymes should be drawn every 1 to 2 weeks initially and then 4 to 6 weeks once a stable dose is achieved. Folate 1 mg daily is recommended for patients on methotrexate (Rheumatrex) to decrease the risk of hepatotoxicity. Other immunosuppressants include azathioprine (Imuran), cyclophosphamide (Cytoxan), and mycophenolate (CellCept). Relapse commonly occurs after stopping immunosuppressants. Currently, no available medications can prevent pulmonary fibrosis (Iannuzzi, 2022).
Other Diffuse Parenchymal Lung Diseases
The fourth category of ILD is termed other DPLD and consists of LAM, PLCH, and eosinophilic pneumonia (King, 2022a).
Lymphangioleiomyomatosis
LAM is a rare, progressive growth of smooth muscle cells throughout the lungs, lymphatics, pulmonary blood vessels, and pleurae that occurs in young women between 20 and 40. The atypical proliferation of smooth muscle cells distorts the lung architecture and leads to cystic emphysema and progressive deterioration of lung function. LAM arises spontaneously, and the cause is unknown, but female sex hormones are hypothesized to play a role. Signs and symptoms include dyspnea and, less commonly, hemoptysis, chest pain, and coughing. Some patients may have crackles or rhonchi, but often there are no other signs of the disease. Many patients present with a spontaneous pneumothorax or a lymphatic obstruction known as chylothorax (i.e., lymph in the digestive system accumulates in the chest cavity). A LAM diagnosis is suspected in young women with dyspnea and interstitial changes, spontaneous pneumothorax, or chylous effusion. HRCT should be done, and findings include multiple, small, diffusely distributed cysts. Serum vascular endothelial growth factor D (VEGF-D) is recommended due to its elevation in most LAM cases. A negative VEGF-D does not exclude LAM, so a surgical lung biopsy is recommended when HRCT and VEGF-D are non-diagnostic. Pulmonary function tests support the diagnosis and typically have an obstructive or mixed pattern. The lungs are usually hyperinflated, and gas trapping is often present. The disease progresses slowly, with a median survival of more than 8 years after diagnosis. Lung transplantation is the standard treatment. Recent research has suggested that sirolimus (Rapamune) treatment can help stabilize or slow the decline in pulmonary function. Sirolimus (Rapamune) should be started at 2 mg daily and adjusted as needed (Lee, 2022j).
Pulmonary Langerhans Cell Histiocytosis
PLCH is a disease characterized by the proliferation of monoclonal Langerhans cells infiltrating the bronchioles and alveolar interstitium, accompanied by plasma cells, eosinophils, neutrophils, and lymphocytes. The cause of PLCH is unknown, but cigarette smoking plays a role, occurring almost exclusively in Whites between 20 and 40 who smoke. Langerhans cell histiocytosis can occur in other organs, but 85% of PLCH cases are exclusively in the lungs. Symptoms of PLCH include fatigue, a nonproductive cough, dyspnea, weight loss, pleuritic chest pain, and fevers, with 15% of patients being asymptomatic. An estimated 10% to 15% of patients with PLCH will have a spontaneous pneumothorax. Physical examination findings are usually normal. Chest radiography shows bilateral, symmetric nodular opacities in the upper and middle lung regions with cystic changes and normal lung volumes. The chest radiograph's appearance can mimic LAM or chronic obstructive pulmonary disease (COPD). The diagnosis of PLCH is confirmed by HRCT with upper and middle lobe cysts or nodules with interstitial thickening. Pulmonary function tests can vary from normal to obstructive, restrictive, or mixed. A reduction in diffusing capacity and impaired exercise are commonly seen. A bronchoscopy and lung biopsy are indicated if HRCT and pulmonary function tests are inconclusive. Spontaneous resolution of symptoms can occur with mild PLCH; the median survival is 8 years. Smoking cessation is the primary treatment, leading to the resolution of symptoms in 33% of cases. Lung transplantation may be an option for otherwise healthy individuals. Corticosteroids and cytotoxic drugs are often used; however, their efficacy is not well established (Lee, 2022o).
Acute Eosinophilic Pneumonia
Acute eosinophilic pneumonia is an acute illness characterized by rapid eosinophilic infiltration of the lung interstitium. It can occur at any age but is most likely between ages 20 and 40 and is twice as prevalent in men than in women. The exact cause is unknown but may be related to an acute hypersensitivity reaction to an unidentified inhaled agent in an otherwise healthy individual. Acute eosinophilic pneumonia presents as an acute febrile illness lasting for fewer than 7 days, with associated dyspnea, myalgias, nonproductive coughing, night sweats, pleuritic chest pain, and malaise. Upon examination, patients may have bibasilar crackles and rhonchi on forced exhalation. These patients may present with acute respiratory failure that requires MV (Lee, 2022a).
A diagnosis of acute eosinophilic pneumonia should be suspected in patients with acute pneumonia that does not respond to antibiotics and progresses to respiratory failure. Other causes of eosinophilic pneumonia (e.g., toxin- or drug-induced, fungal infection) should be ruled out. Unlike chronic eosinophilic pneumonia, a CBC rarely shows markedly elevated eosinophil counts. Chest radiography may show subtle ground-glass or reticular opacities, with opacities that are not characteristically localized to the lung periphery. Small, bilateral pleural effusions occur in up to 66% of cases. HRCT findings will include bilateral, random, patchy reticular or ground-glass opacities. Pulmonary function tests will show a restrictive process with reduced diffusing capacity. Bronchoalveolar lavage should be completed, often showing a high percentage (25%) of eosinophils. Lung biopsy is not frequently done for acute eosinophilic pneumonia. Some patients will improve spontaneously, while others will be treated with corticosteroids. Prednisone (Deltasone) at 40 mg to 60 mg orally daily is the treatment of choice. Methylprednisolone (Solumedrol) at 60 mg to 125 mg IV every 6 hours is preferred for patients with respiratory failure. The response to treatment is usually good, with most patients experiencing complete recovery (Lee, 2022a).
Chronic Eosinophilic Pneumonia
Chronic eosinophilic pneumonia is characterized by a recurrence of acute or subacute eosinophilic pneumonia. Patients experience chronic accumulation of eosinophils in the lungs, which may be related to an allergic diathesis. Patients will present with symptoms that suggest community-acquired pneumonia, including fever, progressive dyspnea, coughing, wheezing, and night sweats. Asthma precedes or accompanies about 50% of cases. Chest radiography findings include bilateral peripheral or pleural opacities commonly in the upper and middle lung regions. HRCT findings are similar to the chest radiograph findings, but the pattern distribution can vary and even be unilateral. Patients with suspected chronic eosinophilic pneumonia should have blood work, including a CBC, erythrocyte sedimentation rate (ESR), and sometimes iron studies. Peripheral blood eosinophilia, a very high ESR, iron-deficiency anemia, and thrombocytosis are often seen with chronic eosinophilic pneumonia. Bronchoalveolar lavage is needed to confirm the diagnosis, with over 40% eosinophilia in the lavage fluid suggesting chronic eosinophilic pneumonia (Lee, 2022c).
Chronic eosinophilic pneumonia is highly responsive to IV or oral corticosteroids. The initial treatment is prednisone (Deltasone) at 40 mg to 60 mg daily. The response to corticosteroids is usually rapid (i.e., within 48 hours), with complete resolution of symptoms and radiograph abnormalities within 14 days. ESR and peripheral eosinophilia counts will also show improvement throughout treatment. However, symptomatic or radiographic relapse occurs frequently after the cessation of corticosteroids; therefore, corticosteroid treatment may be required for years. Inhaled corticosteroids may help reduce the maintenance dose of oral corticosteroids. Fluticasone (Flovent) or beclomethasone (Qvar) at 500 mcg to 750 mcg twice daily are recommended. The chronic relapse of symptoms does not appear to indicate treatment failure or poorer prognosis, and chronic eosinophilic pneumonia is unlikely to cause death (Lee, 2022c).
References
Anatomyclassproj1. (2020). Interstitial lung disease [Image]. https://commons.wikimedia.org/wiki/File:2_SVH_Lung_Health_Interstitial_Lung_Disease_final_1080.jpg
Antoine, M., & Mlika, M. (2022). Interstitial lung disease. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK541084
BruceBlaus. (2013). Respiratory system [Image]. https://commons.wikimedia.org/wiki/File:Blausen_0770_RespiratorySystem_02.png
Cool, C. D. (2021). Idiopathic interstitial pneumonias: Classification and pathology. UpToDate. Retrieved November 30, 2022, from https://www.uptodate.com/contents/idiopathic-interstitial-pneumonias-classification-and-pathology
Hinkle, J. L., & Cheever, K. H. (2018). Textbook of medical-surgical nursing (14th ed.). Wolters Kluwer.
Iannuzzi, M. C. (2022). Sarcoidosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/sarcoidosis/sarcoidosis
King, T. E., Jr. (2022a). Approach to the adult with interstitial lung disease: Clinical evaluation. UpToDate. Retrieved November 30, 2022, from https://www.uptodate.com/contents/approach-to-the-adult-with-interstitial-lung-disease-clinical-evaluation
King, T. E., Jr. (2022b). Hypersensitivity pneumonitis (extrinsic allergic alveolitis): Clinical manifestations and diagnosis. UpToDate. Retrieved November 30, 2022, from https://www.uptodate.com/contents/hypersensitivity-pneumonitis-extrinsic-allergic-alveolitis-clinical-manifestations-and-diagnosis
Lee, J. (2022a). Acute eosinophilic pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/acute-eosinophilic-pneumonia
Lee, J. (2022b). Acute interstitial pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/acute-interstitial-pneumonia
Lee, J. (2022c). Chronic eosinophilic pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/chronic-eosinophilic-pneumonia
Lee, J. (2022d). Cryptogenic organizing pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/cryptogenic-organizing-pneumonia
Lee, J. (2022e). Desquamative interstitial pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/desquamative-interstitial-pneumonia
Lee, J. (2022f). Drug-induced pulmonary disease. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/drug-induced-pulmonary-disease
Lee, J. (2022g). Hypersensitivity pneumonitis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/hypersensitivity-pneumonitis
Lee, J. (2022h). Idiopathic pleuroparenchymal fibroelastosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/idiopathic-pleuroparenchymal-fibroelastosis
Lee, J. (2022i). Idiopathic pulmonary fibrosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/idiopathic-pulmonary-fibrosis
Lee, J. (2022j). Lymphangioleiomyomatosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/lymphangioleiomyomatosis
Lee, J. (2022k). Lymphocytic interstitial pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/lymphocytic-interstitial-pneumonia
Lee, J. (2022l). Nonspecific interstitial pneumonia. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/nonspecific-interstitial-pneumonia
Lee, J. (2022m). Overview of idiopathic interstitial pneumonias. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias
Lee, J. (2022n). Overview of interstitial lung disease. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-interstitial-lung-disease
Lee, J. (2022o). Pulmonary Langerhans cell histiocytosis. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/pulmonary-langerhans-cell-histiocytosis
Lee, J. (2022p). Respiratory bronchiolitis-associated interstitial lung disease. Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/respiratory-bronchiolitis–associated-interstitial-lung-disease
Martinez-Pitre, P. J., Sabbula, B. R., & Cascella, M. (2022). Restrictive lung disease. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK560880
Medic Tests. (2021). Human gas exchange [Image]. https://medictests.com/units/the-mechanics-of-respiration
National Heart, Lung, and Blood Institute. (2022a). Interstitial lung diseases: Causes and risk factors. https://www.nhlbi.nih.gov/health/interstitial-lung-diseases/causes
National Heart, Lung, and Blood Institute. (2022b). Interstitial lung diseases: What are interstitial lung diseases. https://www.nhlbi.nih.gov/health/interstitial-lung-diseases
Norris, T. L. (2020). Porth's essentials of pathophysiology (5th ed.). Wolters Kluwer.